Quantitative PCR (qPCR) is a type of PCR that allows the user to monitor DNA quantity in real time. To do so, qPCR uses a comparative approach, providing quantitative measurements relative to another target, often a reference gene of stable expression.
qPCR is often called real-time PCR, and thus it can be confused with RT-PCR. It is important to note that RT-PCR should only be used for reverse transcription PCR, and the term RT-qPCR indicates reverse transcription qPCR (the most common type of qPCR, as it can be used to measure changes in gene expression).
qPCR uses continuous monitoring of DNA quantity through fluorescent tags, measuring fluorescence intensity after each cycle. The amplification products do not need to be analyzed through an agarose gel, like in the traditional (endpoint) PCR. Because of this, qPCR is a key technique in molecular biology research and even diagnostics.
This article covers the basic principles of how qPCR works, how to set up an experiment and the workflow of running it, and the advantages and applications of the technique over other methods.
Basic Principles of qPCR
qPCR works like traditional PCR. It needs a thermocycler, primers, polymerase, and buffer, and uses amplification cycles similarly. The only difference is that fluorescence is measured after each cycle. With each amplification cycle, the number of target DNA copies present is theoretically doubled. Starting from just a few molecules, this exponential amplification generates billions of copies within a standard run. This gives qPCR its sensitivity even with low-input samples.1

Figure 1: Comparison of traditional PCR vs qPCR, showing instrument examples and readout forms.
The primary output of a qPCR run is the Cq value (quantification cycle) of each sample/well (also known as Ct, or threshold cycle). This is the cycle at which the fluorescent signal first crosses a defined background threshold. A lower Cq means more starting material was present: a sample with 10,000 copies will amplify detectably earlier than one with 100, and thus have a lower Cq.2
When it comes to detecting fluorescence, there are two main types of molecules:
- Intercalating dyes like SYBR Green bind to any double-stranded DNA produced during amplification. These are cost-effective and broadly applicable, but unable to distinguish target amplicons from primer dimers.
- Sequence-specific hydrolysis probes like TaqMan only generate a signal when cleaved at the specific target sequence, offering higher specificity at a higher cost. For most labs and routine gene expression work, SYBR Green is often enough, but for rare target detection or multiplexing, probes are the better choice.3
Typical Workflow
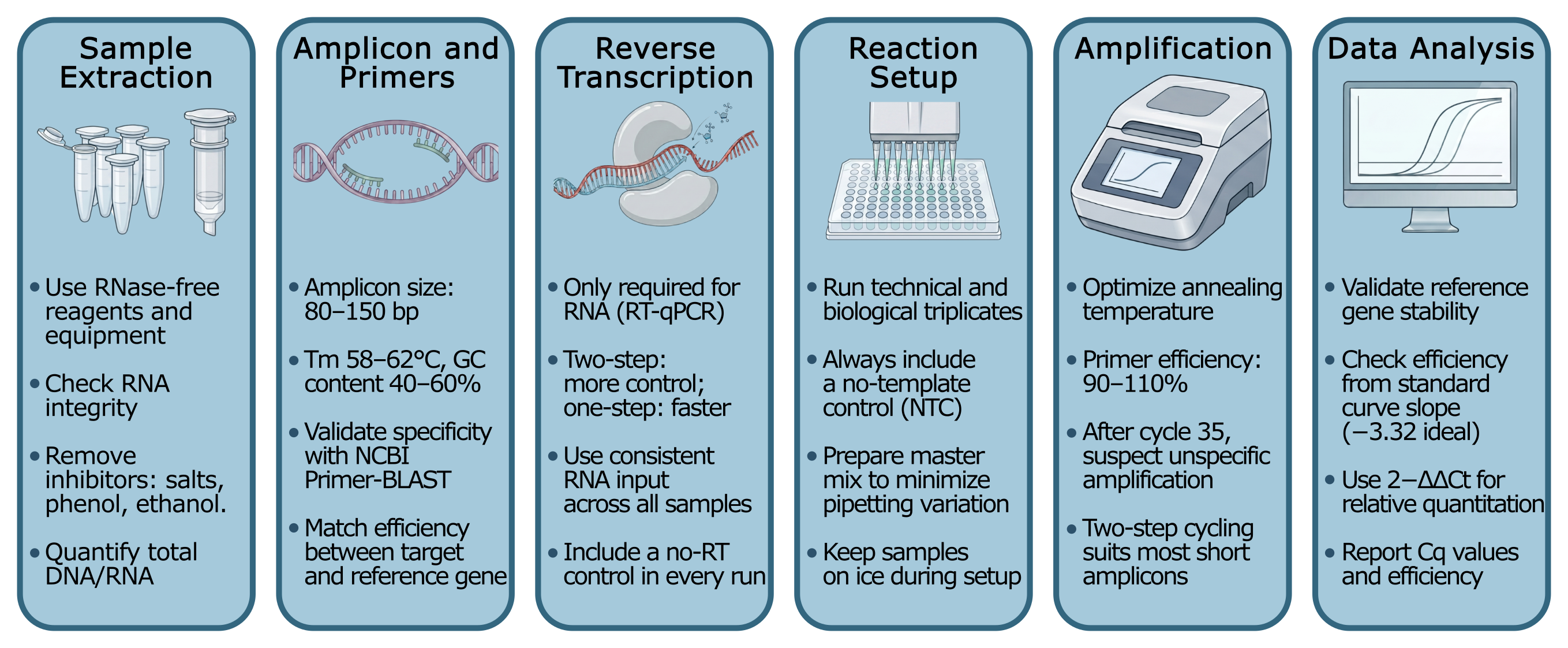
A standard qPCR experiment is more complex than most people think. In this section, you will find a general overview, but know there is much more to each step. If you are dealing with problems or want more in-depth data, you can check the MIQE guidelines4 or look for protocol optimization papers and options for your specific use case.
A core issue with qPCR is that errors compound, as it relies on exponential amplification. Small errors in each step lead to big detection shifts, and differences in amplification efficiency between target and reference genes introduce bias. When this happens, comparison between target and reference gene quantities do not reflect real biological variations.
Sample preparation and nucleic acid extraction
The reaction is only as good as the input material. RNA and DNA need to be pure and free of inhibitors like phenol, ethanol, or excess salts. These interfere with polymerase activity. For RNA, integrity matters too: degraded RNA results in underestimated expression values and poor reproducibility.5
Target and primer design
Before running a single reaction, primer design and validation deserve serious attention. Primers should be 18–22 bp long, have a melting temperature (Tm) between 58–62°C, a GC content of 40–60 %, and produce an amplicon of 80–150 bp. Tools like NCBI Primer-BLAST can check specificity against the genome and flag off-target binding sites.
Efficiency is critical in comparative quantitation. A well-designed primer pair should amplify with ~100 % efficiency, meaning the template nearly doubles with every cycle. Efficiency is measured by running a standard curve across a serial dilution (typically 5 logs) and calculating it from the slope: an ideal slope of −3.32 corresponds to 100 % efficiency (3.32 cycles to 10x DNA quantity), and the acceptable range is 90–110 %. If your target and reference gene do not amplify with similar efficiency, the analysis and interpretation will be incorrect.6
Reverse transcription (for RT-qPCR)
If you're working with RNA, it needs to be transcribed to cDNA. This is done by reverse transcriptase before amplification. You can run this as a two-step assay (RT first, qPCR second) for flexibility. Or, you can use a one-step assay where both reactions happen in the same tube/well, which is faster and more convenient, but offers less control over reaction conditions.7
Reaction setup
Each well gets a mix containing: the DNA template (or cDNA), target-specific primers (and probe if applicable), a thermostable DNA polymerase, buffer, dNTPs, and the fluorescent molecules. Running both technical replicates (usually triplicates) and proper controls is crucial.4 Controls include a no-template control (NTC) to catch contamination, and a no-reverse-transcriptase control (no-RT) for RNA work.
Thermal cycling
The plate runs through repeated cycles of denaturation (~95 °C), annealing, and extension. The instrument reads fluorescence at the end of each cycle, building the amplification curve in real time. Most protocols run 40 cycles, but anything amplifying after cycle 35 is typically considered background/non-specific amplification.8 Annealing temperature, two-step vs three-step cycling (annealing+extension together or separate), and cycle speed/length all influence your amplification efficiency.
Data analysis
Software uses the amplification curves to calculate Cq values for each well. From there, you can run quantification against a standard curve of known concentrations or relative quantification comparing target expression to a reference gene using the 2−ΔΔCt method. Assay efficiency can also be calculated from the standard curve slope and needs to be validated before drawing biological conclusions.6
Figure 2: A visual representation of a step-by-step qPCR workflow, showing the 6 steps outlined before.
Advantages of of qPCR
qPCR has largely replaced gel-based endpoint PCR in research and diagnostic settings as it delivers quantitative data faster and with less error.9
Workflow and performance
Thanks to real/time detection, post-PCR processing is eliminated. The plate is never opened, which also removes carry-over contamination, a persistent problem in gel-based workflows.4,10 A standard run, including analysis, takes 2–3 hours from extracted nucleic acid to readout.
Sensitivity and dynamic range
qPCR detects targets down to single-copy level and maintains linear quantification across 6–7 orders of magnitude. The same assay reliably measures abundant housekeeping genes and rare transcripts without redesign.5 That said, sensitivity is dependent on assay design and optimization.
Multiplexing
New instruments support 4–6 simultaneous target detection using spectrally distinct fluorophores, reducing sample consumption and run time. Probe-based multiplexing also allows co-detection of internal positive controls in the same reaction well, improving data reliability without adding runs.11 Newer approaches like color cycle multiplexing are pushing that ceiling further, detecting 21 targets in a single tube.12
Practical considerations
To run qPCR experiments, you’ll need to purchase a machine. The cost of this varies wildly, and researchers must account for their use. Entry-level instruments with fewer fluorescence channels and fixed block formats limit multiplexing and throughput, and often have a higher cost per reaction. However, the upfront cost can be only a few $1,000s. 384-well cyclers that allow for multiplexing are better for high throughput, and require less reagent volume (and thus, less cost/reaction), but can hit $30,000+. Maintenance, support, and reagents matter as much as quality when choosing a machine. Reaction costs are around $0.5–1 for single targets, and up to $2.5 if multiplexing.
Applications of of qPCR
qPCR is used across basic research, clinical diagnostics, and translational medicine, as few techniques match its combination of sensitivity, speed, and output at the cost qPCR has.
Research and molecular biology
RT-qPCR is the standard method for quantifying gene expression and validating findings from discovery-scale experiments. When RNA-seq identifies differentially expressed genes, RT-qPCR is used to confirm those differences. It is also routinely used for genotyping and copy number analysis, where quantifying the number of copies of a sequence matters as much as the presence of the sequence.13
Clinical diagnostics
In infectious disease, several qPCR-based assays hold full FDA approval or CE-IVD certification for clinical use. qPCR platforms from manufacturers including Roche, Abbott, bioMérieux, and QIAGEN are certified for quantifying viral loads of HIV, HCV, CMV, and other high-burden pathogens. These are used routinely to confirm infection, stratify patients, and monitor treatment response. During the COVID-19 pandemic, RT-qPCR was the primary globally deployed diagnostic. The widespread use of RT-qPCR during the pandemic has shaped how molecular diagnostic performance standards are now defined.14
Oncology
In oncology, qPCR confirms mutations identified by NGS, quantifies gene expression of drug targets such as EGFR or HER2 in tissue, and measures treatment response at the transcript level. For applications requiring absolute quantification of rare variants or ctDNA at very low allele frequencies, digital PCR offers better precision, but qPCR remains the accessible, high-throughput workhorse for expression-based biomarker work where relative quantification is sufficient.
Frequently Asked Questions (FAQs)
1. What is the difference between qPCR, RT-PCR, and RT-qPCR?
Quantitative PCR (qPCR) is also known as real-time PCR. RT-PCR stands for reverse transcription PCR. RT-qPCR is reverse transcription quantitative PCR. “RT” always stands for “reverse transcription” and not “real time.”
2. What are Ct and Cq values, and what are acceptable values?
Ct value = Cq value. Ct stands for threshold cycle, Cq for quantification cycle, but they refer to the same number. There is no universal "good" Ct/Cq value, they change based on starting target DNA. Most reliable results fall between cycles 15 and 35, and the lower the value, the more starting target DNA. A Cq above 35 is typically considered background or non-specific amplification and should be treated with caution.
3. How do I know if my qPCR efficiency is acceptable?
Run a standard curve across a 5-log serial dilution and calculate efficiency from the slope. An ideal slope of −3.32 corresponds to 100% efficiency, and the acceptable range is 90–110%. If your target and reference gene differ significantly in efficiency, your relative quantification results will be unreliable.
References
1. Kubista M, Andrade JM, et al. The real-time polymerase chain reaction. Mol Aspects Med. 2006;27(2–3):95–125. https://doi.org/10.1016/j.mam.2005.12.007
2. Ruijter JM, Ramakers C, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37(6):e45. https://doi.org/10.1093/nar/gkp045
3. Navarro E, Serrano-Heras G, et al. Real-time PCR detection chemistry. Clin Chim Acta. 2015;439:231–250. https://doi.org/10.1016/j.cca.2014.10.017
4. Bustin SA, Ruijter JM, et al. MIQE 2.0: Revision of the Minimum Information for Publication of Quantitative Real-Time PCR Experiments Guidelines. Clin Chem. 2025;71(6):634–651. https://doi.org/10.1093/clinchem/hvaf043
5. Taylor SC, Nadeau K, et al. The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends Biotechnol. 2019;37(7):761–774.
https://doi.org/10.1016/j.tibtech.2018.12.002
6. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. https://doi.org/10.1006/meth.2001.1262
7. Wong ML, Medrano JF. Real-time PCR for mRNA quantitation. BioTechniques. 2005;39(1):75–85. https://doi.org/10.2144/05391rv01
8. ue B, Zhang H, et al. A TaqMan qPCR for precise detection and quantification of diarrheagenic Escherichia coli. Sci Rep. 2025;15:16728. https://doi.org/10.1038/s41598-025-96960-1
9. Valasek, M. A., & Repa, J. J. (2005). The power of real-time PCR. Advances in Physiology Education, 29(3), 151–159. https://doi.org/10.1152/advan.00019.2005
10. Espy MJ, Uhl JR, et al. Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin Microbiol Rev. 2006;19(1):165–256. https://doi.org/10.1128/CMR.19.1.165-256.2006
11. Zhang H, Yan Z, et al. Determination of Advantages and Limitations of qPCR Duplexing in a Single Fluorescent Channel. ACS Omega. 2021;6(34):22292–22300. https://doi.org/10.1021/acsomega.1c02971
12. Chen W, Zhang K, et al. Advancing quantitative PCR with color cycle multiplex amplification. Nucleic Acids Res. 2024;52(17):e81. https://doi.org/10.1093/nar/gkae683
13. Aguiar ERGR, Almeida FL, et al. Comparison between qPCR and RNA-seq reveals challenges of low expression quantification. Immunogenetics. 2023;75:175–185. https://doi.org/10.1007/s00251-023-01296-7
14. Bustin, S. A. (2024). RT-qPCR Testing and Performance Metrics in the COVID-19 Era. International Journal of Molecular Sciences, 25(17), 9326. https://doi.org/10.3390/ijms25179326