Statistics and Interpretation
The last thing to do is to correct the data to obtain the absolute number of copies. The most common way to calculate copies per microliter is to use the Poisson correction.7 However, other methods have gained some traction in the last few years. Lastly, additional considerations must be taken into account when analyzing multiplex dPCR data.
Absolute quantification using Poisson correction
Digital PCR’s power derives from binary endpoint detection: each partition scores as positive (1) or negative (0) after PCR, independent of amplification efficiency.
Molecules are assumed to distribute randomly among partitions, following Poisson statistics, so the fraction of negative partitions (p₀) directly relates to the average molecules per partition (λ). But if 8,000 partitions are positive out of 20,000, that does not mean there are 8,000 target molecules, as some partitions may have 2, 3, or more copies.
The fraction of negative partitions follows a Poisson distribution. The average molecules per partition (λ) is calculated as:
λ = −ln(p₀) or equivalently λ = −ln(1−p)
Where p₀ = fraction of negative partitions (negative partitions/ total partitions)
Total copies per reaction = λ × (total partitions)
Concentration = Total copies / reaction volume (in µL) = copies/µL
Example: If 8,000 of 20,000 partitions are positive (40% positive, 60% negative):
- λ = −ln(0.60) = 0.51 copies per partition
- Total copies = 0.51 × 20,000 = 10,200 copies<
- Concentration = 10,200 / 20 µL = 510 copies/µL
Statistical precision improves with more partitions: the coefficient of variation decreases with partition number, meaning 20,000 partitions yield ~0.7 % theoretical precision, while 10,000 partitions provide ~1 %.
95 % confidence intervals are calculated from the binomial distribution of positive/negative counts, widening as positive fractions approach saturation (>95 %) or scarcity (<1 %).
Unsure about how to set up a good experiment to obtain good confidence intervals? Our guide on dPCR assay design gives you practical tips to do so.
Alternative statistical methods
Standard dPCR quantification assumes random distribution of molecules (Poisson distribution) and identical partition volumes, but these assumptions can be violated (like partition volume differences, non‑random loading, etc.).8
When partition volumes vary, pure Poisson models become biased at higher concentrations, and extended models like Poisson-Plus have been developed to help with that.9 Other, more flexible approaches can provide higher accuracy in certain circumstances.
For uncertainty estimation, several papers explicitly model the number of positive partitions as binomial (or multinomial in multiplex) and develop methods (delta method, binomial bootstrap “BinomVar”, “NonPVar”, etc.) that work on the discrete positive/negative counts instead of assuming an ideal infinite‑partition Poisson process.10
In parallel, generalized linear (mixed) models with complementary log-log links have been proposed. These embed the Poisson model in a GLM framework, allowing partition volume to be included as an offset, and experimental factors (such as assay, replicate, or treatment) to entered as covariates. This improves variance estimates when additional error sources are present.11
For routine applications where partitioning is approximately random and technical variability is limited, standard Poisson‑based quantification is generally adequate. However, if precise uncertainty quantification or method validation is required, other models provide more flexible (and often more accurate) variance estimates than simple Poisson formulas.
For standardized dPCR data analysis and reporting, follow the dMIQE 2020 guidelines, the accepted community standard for digital PCR experiment reporting.6
Multiplexing considerations
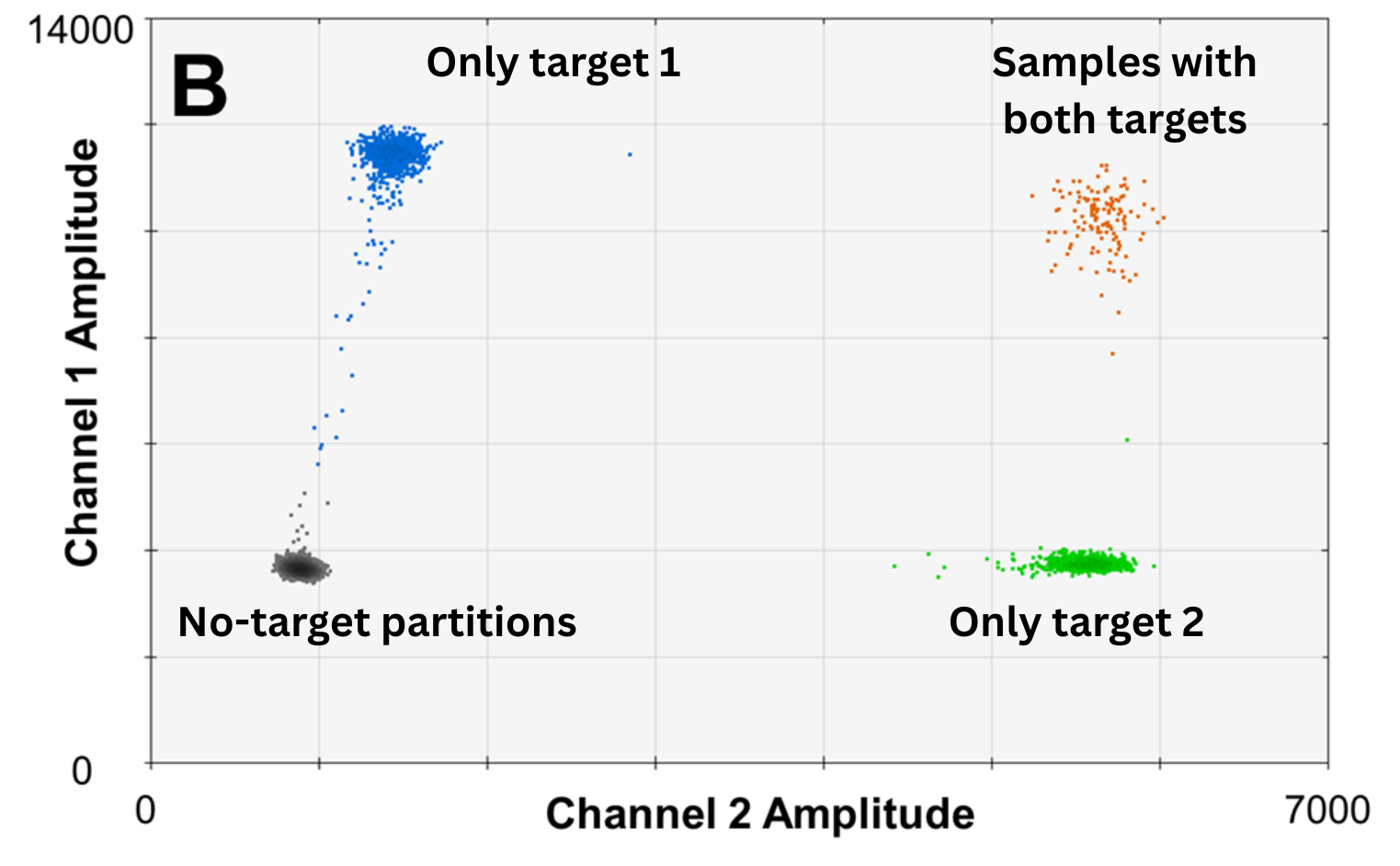
Figure 3. Example of output of multiplex dPCR with two targets. Dots represent partitions with none, one, or two targets. Note that the rain phenomenon is still visible. Modified from Hughesman CB, Lu XJD, et al. PLoS One. 2016;11(8):e0161274, Supplementary Figure 2 B.12
Multiplexed dPCR requires mathematical deconvolution to separate overlapping fluorescence signals. Spillover compensation matrices correct spectral bleed-through between channels, particularly critical in six- to seven-color systems.
The analysis involves solving linear equations where total fluorescence in each channel equals the sum of individual fluorophore contributions multiplied by their spectral overlap coefficients.13
Reference gene normalization in multiplex CNV assays requires propagation of uncertainty from both numerator and denominator, compounding confidence intervals.14
Remember to always validate multiplex performance by comparing single-plex versus multiplex results. Significant deviation indicates primer competition or probe cross-reactivity requiring assay redesign.
This article is part of a 3-part series on digital PCR. Continue reading: