Massachusetts researchers have developed a strategy to treat two of the most common inherited blood diseases—sickle cell disease and beta thalassemia—applying CRISPR-Cas9 gene editing to patients' own blood stem cells. Described in two papers, one published this week in Nature Medicine and the other earlier this month in Blood, their approach reportedly overcomes prior technical challenges, editing blood stem cells more efficiently than in the past.
"We think our work defines a strategy that could lead to the cure of common hemoglobin disorders," says Daniel Bauer, M.D., Ph.D., an attending physician with Dana-Farber/Boston Children's and a senior author on both papers. "Combining gene editing with an autologous stem-cell transplant could be a therapy for sickle-cell disease, beta-thalassemia and other blood disorders."
The Nature Medicine study used CRISPR-Cas9 technology, in particular a Cas9 protein modified by a team led by Scot Wolfe, Ph.D., at UMass Medical School, to optimize gene editing. In previous attempts to edit the genomes of human blood stem and progenitor cells, the efficiency, specificity and long-term stability of the edits once the cells engraft in the bone marrow have varied. The new technique improves the targeting and durability of the edits.
Search Antibodies Search Now Use our Antibody Search Tool to find the right antibody for your research. Filter
by Type, Application, Reactivity, Host, Clonality, Conjugate/Tag, and Isotype.
Bauer's team used the strategy to make a highly targeted edit. Previous work at Boston Children's had showed that inactivating a gene called BCL11A allows red blood cells to keep producing a fetal form of hemoglobin even after birth. Fetal hemoglobin doesn't sickle and can stand in for defective "adult" hemoglobin. More recently, Bauer found a safer target: a genetic enhancer of BCL11A that is active only in red blood cells.
"With our new very efficient protocol, we can edit the BCL11A enhancer in nearly all blood stem cells we collect, overcoming some of the technical challenges of editing these cells," says Bauer. "In our experiments, more than 95 percent of copies of the enhancer sequence were changed in a way we expect would be therapeutic."
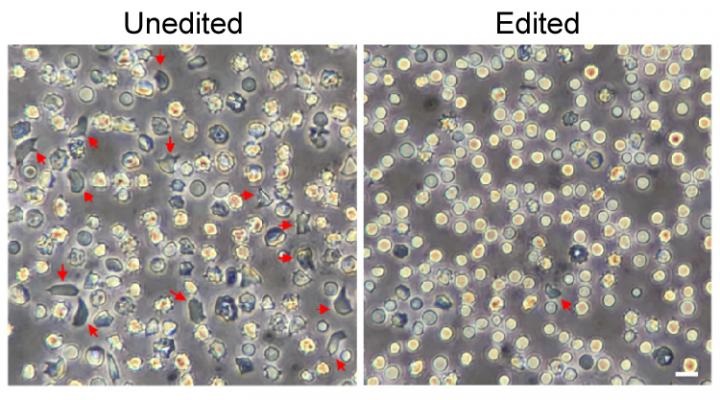
The strategy enabled mice carrying blood stem cells from patients with sickle cell disease to produce red blood cells with enough fetal hemoglobin to prevent cell sickling. The team showed that the gene-edited cells, infused back into the bloodstream, engrafted in the bone marrow and produced genetically corrected red blood cells. Later, when blood stem cells were isolated from these mice and transplanted into other mice, the cells engrafted again, still carrying the therapeutic gene changes.
Applied to blood stem cells from patients with beta-thalassemia, the same strategy restored the normal balance of the globin chains that make up hemoglobin.
The other study, published in Blood, used a similar gene editing protocol to target forms of beta-thalassemia that involve splicing mutations—errors in bits of DNA near the beta-globin gene that change how the gene is read out to assemble beta-globin protein. In this study, nine patients with beta thalassemia donated their cells, which were manipulated in a dish. For some patients, the UMass team produced a different enzyme, Cas12a, to more effectively target their mutations. The CRISPR system efficiently made edits and restored normal splicing of the beta-globin protein in blood cells from each of the patients.
Image: The unedited red blood cells at left include many misshapen 'sickle' cells, shown by red arrows. These largely disappeared in the red blood cells made by gene-edited blood stem cells. Image courtesy of Daniel Bauer/Boston Children's Hospital.