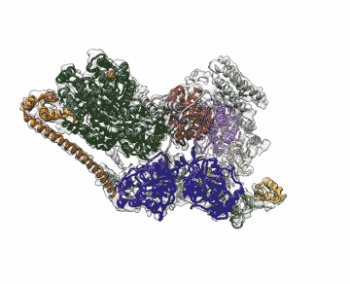
Researchers at the DOE’s Lawrence Berkeley National Laboratory (Berkeley Lab) created what they say is the highest resolution map yet of a large assembly of human proteins that are critical to DNA function. Specifically, they used cryo-electron microscopy (cryo-EM) to resolve the 3D structure of transcription factor IIH (TFIIH) at 4.4 angstroms, or near-atomic resolution.
TFIIH is important as it is used to unzip the DNA double helix so that genes can be accessed and read during transcription or repair.
"When TFIIH goes wrong, DNA repair can't occur, and that malfunction is associated with severe cancer propensity, premature aging, and a variety of other defects," said study principal investigator Eva Nogales, faculty scientist at Berkeley Lab's Molecular Biophysics and Integrated Bioimaging Division. "Using this structure, we can now begin to place mutations in context to better understand why they give rise to misbehavior in cells."
"As organisms get more complex, these proteins do too, taking on extra bits and pieces needed for regulatory functions at many different levels," added Nogales, who is also a UC Berkeley professor of molecular and cell biology and a HHMI investigator. "The fact that we resolved this protein structure from human cells makes this even more relevant to disease research. There's no need to extrapolate the protein's function based upon how it works in other organisms."
Biomolecules are typically imaged using X-ray crystallography, but that method requires a large amount of stable sample for the crystallization process to work. The challenge with TFIIH is that it is hard to produce and purify in large enough quantities, which is one of the reasons the team relied on cryo-EM, which can work even when sample amounts are very small.
According to the team, cryo-EM was ideal for this project because of advanced detector technology that Berkeley Lab engineer Peter Denes helped develop. Instead of a single picture taken for each sample, the direct detector camera shoots multiple frames. The frames are then put together to create a high-resolution image. This approach resolves the blur from sample movement. The improved images contain higher quality data, and they allow researchers to study the sample in multiple states, as they exist in the cell.
This approach, however, generates copious amounts of data, such that the supercomputers at the National Energy Research Scientific Computing Center (NERSC) at Berkeley Lab had to be used to process all of it.
"When we began the data processing, we had 1.5 million images of individual molecules to sort through," said Greber. "We needed to select particles that are representative of an intact complex. After 300,000 CPU hours at NERSC, we ended up with 120,000 images of individual particles that were used to compute the 3D map of the protein."
To obtain an atomic model of the protein complex based on this 3D map, the researchers used PHENIX (Python-based Hierarchical ENvironment for Integrated Xtallography), a software program whose development is led by Paul Adams, director of Berkeley Lab's Molecular Biophysics and Integrated Bioimaging Division and a co-author of this study.
Not only does this structure help with basic understanding of DNA repair, the information could be used to help visualize how specific molecules are binding to target proteins in drug development.
Image: The cryo-EM structure of Human Transcription Factor II (TFIIH). The atomic coordinate model, colored according to the different TFIIH subunits, is shown inside the semi-transparent cryo-EM map. Image courtesy of Basil Greber/Berkeley Lab and UC Berkeley.