Beyond knowing the sequence of DNA, scientists want to know what it’s doing. To find out, many researchers turn to transcriptome analysis. Sometimes that means looking at single cells. Most important, scientists need various ways—reliable and easy methods—for analyzing what is being transcribed and when.
When asked about some of the key applications of transcriptome analysis, Pete Hedley, genome technology group leader at The James Hutton Institute, says, “We utilize transcriptomics for high-throughput, gene-expression analysis in crop species—barley, potato, and soft fruit—along with associated pathogens.” Hedley and his colleagues do this to help identify genes that could impact the quality and resilience of the crops. “In addition, we have interest in RNA isoform analysis and alternative splicing,” he says.
The applications of transcriptome analysis depend largely on the available methods and products that can be used.
The single-cell attack
In 2013, Jason Buenrostro—then a doctoral student at the Stanford University School of Medicine and now an assistant professor of stem cell and regenerative biology at Harvard University—and his colleagues described in Nature Medicine an assay for transposase-accessible chromatin using sequencing (ATAC-seq). In describing the potential of this assay, the team wrote: “Using ATAC-seq maps of human CD4(+) T cells from a proband obtained on consecutive days, we demonstrated the feasibility of analyzing an individual’s epigenome on a timescale compatible with clinical decision-making.”
Based on this assay, Bio-Rad developed a new single-cell ATAC-Seq solution. “This is a complete next-generation sequencing library prep solution for single-cell ATAC-Seq with flexible cell throughput, high sensitivity, capture efficiency of 50% or greater, and a single-day workflow,” says Carolyn Reifsnyder, director of life science marketing for the digital biology group at Bio-Rad.
The high sensitivity means obtaining more unique fragments that map to transcription start sites in the nuclear genome. “You can profile epigenomic patterns at the single-cell level to better understand the mechanisms that drive which genes are turned on and off,” Reifsnyder says. This assay maps a cell’s open chromatin. As Reifsnyder explains, “The data is used to infer regions of increased or decreased accessibility.”
To use this assay, users start with a suspension of single cells and then extract the nuclei. Next, Bio-Rad’s ddSEQ Single-Cell Isolator encapsulates single nuclei and barcoded beads into droplets. Finally, library preparation and sequencing occur.
“The method doesn’t require that you know which sites you need to study,” Reifsnyder says. “Rather it lets you map the landscape across a single cell and then aggregate data for hundreds to thousands of single cells.” Just as important, only a couple hundred cells can be enough.
Initially, this assay is being adopted primarily in epigenetics research, and will move into the broader research and translational research fields. In oncology, for example, this assay can provide insight into the earliest stages of dysregulation among cells. In stem-cell and developmental biology, scientists can use this tool to resolve dynamic changes in chromatin during normal embryogenesis. In neurobiology, single-cell sequencing might help scientists correlate changes in the transcriptome with a diseases state.
“We’re just seeing the tip of the iceberg of what people can do with this technology,” Reifsnyder says.
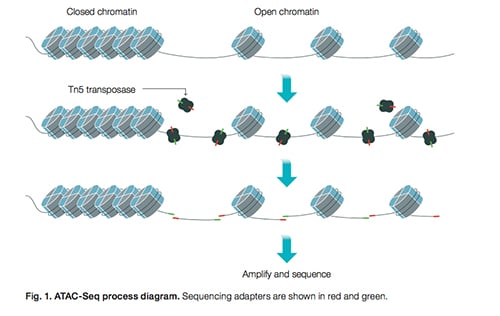
Image: The ATAC-Seq process is an assay for transposase-accessible chromatin using sequencing. Image courtesy of Bio-Rad.
Understanding un-fragmented analysis
Transcriptome analysis can be done on fragmented or un-fragmented stretches of nucleic acids. “Un-fragmented transcriptome analysis consists of sequencing cDNA using long-read sequencing technology,” says Laurence Ettwiller, senior scientist in the research department of New England Biolabs. She compares this to using RNA-seq in fragmented transcriptome analysis where long sequencing reads identify transcripts, and the number of reads per transcript correlates with the level of transcript in the cells. “Unlike RNA-seq,” Ettwiller points out, every read from an “un-fragmented transcriptome should ideally correspond to a full length transcript, revealing the structure of the operons.”
With SMRT-Cappable-seq from New England Biolabs, scientists can explore the prokaryotic transcriptome without fragmentation. “By removing process transcripts, which correspond to the vast majority of transcripts, SMRT-Cappable-seq allows for the sequencing of only primary transcripts, thus revealing the correct operon structure,” Ettwiller explains. “SMRT-Cappable-seq combines the isolation of prokaryotic primary transcripts with long read–sequencing technology.”
Isolating the primary transcripts starts with labeling the 5'- and 3'-end of un-fragmented prokaryotic primary transcripts. Then, labeled primary transcripts are enriched and converted into cDNAs. Last, the entire cDNA molecules are amplified and sequenced using long-read sequencing technology from Pacific Biosciences.
This company keeps providing new options in transcriptome analysis. “PacBio’s latest Sequel chemistry provides both long and accurate sequence reads, thereby generating full-length, high-fidelity transcript reads,” says Jason Underwood, principal scientist at Pacific Biosciences. “This power is further combined with a new tool called IsoPhase, which can distinguish whether gene products originate from the maternal and paternal genome copy—leveraging sequence differences between the two alleles.” With this full-length transcript phasing, scientists can run experiments that reveal allele-specific isoform expression at higher resolution and across broader scales.
Others agree on the importance of the PacBio technology. When asked about the most interesting recent technological advance related to transcriptome analysis, Hedley mentions “long-read technologies for transcriptomics, specifically Pacific Biosciences Iso-Seq for identification of full-length transcript isoforms.”
RNA-seq is currently the gold-standard technology for quantifying gene expression on a genome-wide scale. “Because of the necessary fragmentation inherent to short-read technology, current methods for characterizing transcriptomes, such as RNA-seq, provide an imperfect characterization of transcriptional landmarks such as transcription start sites, termination sites, and operon structure,” Ettwiller says. “Un-fragmented transcriptome analysis, on the other hand, provides a more complete picture of where the transcription starts and ends, as well as the gene composition of full length operons.”
Without needing to assemble sequence fragments, this approach provides other opportunities. “It could be applied to novel microbiomes without genome sequencing or gene annotation,” Ettwiller says. “Furthermore, long-read sequencing directly provides the information of genes contained in the same operon, which facilitates the identification of various metabolic pathways.” Currently, scientists at New England Biolabs are adapting SMRT-Cappable-seq to nanopore sequencing and extending the application to also include eukaryotic full-length transcriptomes.
For new research techniques, including single-cell ATAC-Seq and SMRT-Cappable-seq, many more applications and discoveries lie ahead. “Single-cell NGS is still a rapidly growing market,” Reifsnyder says. A report from Grand View Research forecasts that the “global single-cell genome sequencing market size is expected to reach USD 2.49 billion by 2025.” That’s a big market, and a big-market forecast comes from high expectations. Certainly, these technologies will help researchers better understand the molecular mechanisms behind healthy and diseased processes.