Abstract
Label-free technologies are emerging technology platforms which have recently been introduced into the pharmaceutical industry for applications such as examining GPCR signaling. In this paper, we will discuss how label-free technology platforms can be selectively integrated to support GPCR drug discovery processes. Utilities of label-free technology platforms from target validation to lead optimization were addressed through several case studies. Advantages of implementing label-free technology platforms were also demonstrated in a few examples of GPCR mechanistic analysis as well as structure activity relationship (SAR) studies.
Introduction
Drug discovery processes used in the 1970s were mainly driven by limited random screening and serendipity. These processes were strengthened considerably during the 1980s to allow for a more rational approach to structure–activity relationship studies. By the 21st century, the drug discovery processes were accelerated significantly due to advanced technology implementation. Evolution of drug discovery processes ultimately led to process standardization across industry. As a result, the drug discovery process can be divided into four critical components including target ID/validation, hit identification, hit assessment and lead optimization (see Figure 1). Therefore, it is not surprising that the success of drug discovery programs depends highly upon how to effectively integrate these four components and subsequently maximize the values that are delivered from each component.
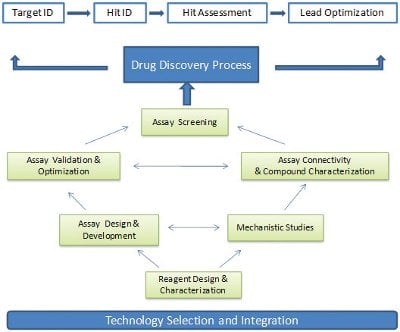
Figure 1- Technology selection and integration are essential for executing assay screening strategy and driving a highquality drug discovery process.
In our opinion, the primary goal of drug discovery process integration is to build the best connectivity from one step to another and to eliminate resource waste throughout the entire process. Although numerous activities are involved in each discovery process component, one of the key activities, assay development and execution, is obviously needed throughout the drug discovery processes. The results derived from relevant assays at each step directly impact the connectivity between steps. Technology selection is the key to determine the quality of assay execution strategy. Therefore, integration of technology platforms across discovery processes became an essential mission to transfer knowledge from one step to another.
Assay technology integration is dynamic and requires continuous adaptation of diverse results from different technology platforms. The rationale behind this notion is four fold. First, the options of assay technology platforms at each stage of the drug discovery process are often more than one (i.e., biochemical assays vs. cell-based assays). Second, the need to emphasize assay throughput, assay sensitivity and assay dynamic range may vary from the early drug discovery process (i.e., hit identification) to the late drug discovery process (i.e., lead optimization). Third, biological systems can be completely different at each discovery component (i.e., recombinant system vs. physiologically relevant systems). Finally, prior to implementing an assay screening plan, a series of key activities are required to ensure the quality of assay screening results. These activities interrogate each other and form a complex network to deliver countable results to meet the needs of discovery programs (Figure 1).
Technology solutions have been proven to be capable of delivering key milestones along drug discovery processes. In the past few years, a rapid growing technology field driven by various label-free technology platforms began to draw attention across industry due to their unique applications and flexible functions [1-9]. In this paper, we will use G-protein coupled receptor (GPCR) drug discovery processes as a template to discuss how to selectively integrate label-free assay technology platforms to impact each critical decision point. From these case studies, we conclude that the utilities of emerging label-free technology platforms can be broadly applied at the frontier of GPCR drug discovery and that selective integration of such platforms with label technology platforms are essential to drive the best value of these technology solutions.
Materials and Methods
Compounds and Peptides
Glucagon-like peptide-1, dopamine HCl, carbachol and pertussis toxin were obtained. MCP-1 and MIP-1α were purchased. Dopamine HCL and acetylcholine were prepared as stocks in water. Pertussis toxin was dissolved in 50% (v/v) glycerol containing 50 mM Tris, pH 7.5, 10 mM glycine, and 0.5 M sodium chloride. Glucagon-like peptide-1, MCP-1 and MIP-1α were dissolved in phosphate buffered saline.
Cell Culture
Chinese hamster ovary (CHO) cells stably expressing the human GLP-1 receptor were cultured in Ham’s F-12 media containing 10% heat inactivated fetal bovine serum and 400μg/ml aminoglycoside antibiotic. CHO cells stably expressing the dopamine D2s receptor were cultured in MEM alpha media containing 10% heat inactivated fetal bovine serum and 600μ/ml aminoglycoside antibiotic. CHO cells stably expressing the muscarinic M1 receptor were grown in Ham’s F-12 media containing 10% qualified fetal bovine serum, 1% sodium pyruvate and 200 μg/ml aminoglycoside antibiotic. CHO cells stably expressing a Gq-coupled GPCRX were cultured in Ham’s F12 media containing 5% dialyzed fetal bovine serum, 5% charcoal/dextran fetal bovine serum, 1X MEM Non-essential Amino Acids and 500μg/ ml aminoglycoside antibiotic and 250μg/ ml Zeocin. CHO cells stably expressing a Gi-coupled GPCRx for SAR study were grown in Ham’s F-12 media containing 10% qualified fetal bovine serum, 1% sodium pyruvate and 500 μg/ml aminoglycoside antibiotic. Human acute monocytic leukemia THP-1 cells were maintained in RPMI 1640 media containing 10% fetal bovine serum, 1% penicillin/streptomycin and 55μM 2-mercaptoethanol.
For cryopreservation experiments, GPCRX expressing CHO cells were harvested using reagent, centrifuged at 1000 rpm for 5 minutes and the cell pellet was suspended in 90% FBS and 10% DMSO, frozen at -80°C overnight and transitioned to a liquid nitrogen tank. Cells are thawed at time of experiment by gentle agitation in a 37°C water bath and transferred to 12 ml of complete growth media for washing and resuspension.
Label-free Assays
Automated microscopy was applied for real-time tracking of A875 human melanoma cell morphology and growth up to 124 hours. Cells were plated at three different densities in a 384-well black clearbottom cell culture plates: 300 cells/well, 500 cells/well and 800 cells/well, separately. The cellular proliferation assay with compound treatment starts 16 hours after cells were plated. After the cells received compound treatment, the effects of compounds are measured by MTS (3-(4,5-dimethylthiazol- 2-yl)-5-(3-carboxymethoxyphenyl)-2-(4- sulfophenyl)-2H-tetrazolium) cytotoxicity assays at the 90 hour time point.
For CHO cell lines, cells were dissociated with trypsin and plated at a final density of 10,000 cells/well in a 384-well microtiter plate in 30 μl of cell growth media. The plate was centrifuged briefly (100rpm for 1 minute) to remove air bubbles. The cell plates were then incubated overnight in a tissue culture incubator at 37ºC in 5% CO2. The next morning, cell growth media was exchanged with 20 μl/well assay buffer, 20 mM HEPES, 0.1% BSA, 0.5% DMSO using online fluidics and incubated at 37ºC for 45 minutes on the instrument. A baseline reading was established for 15 minutes followed by fluidics addition of 10 μl of 3X compound/agonist in assay buffer. Impedance responses were measured for 30 minutes. For cryopreserved studies for GPCRX, frozen cell vials were thawed at the time of plating followed by assay the following morning.
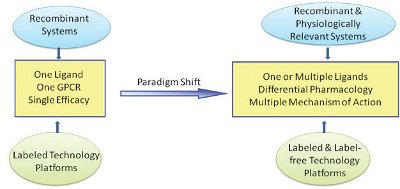
Figure 2- Label-free technology platforms in conjunction with labeled technology platforms lay out the key foundation to understand GPCR drug discovery programs from target identification to lead optimization.
For suspension THP-1 human acute monocytic leukemia cells, the cells were spun down at 1000 rpm for 3 minutes and washed once with wash buffer (1X Hanks buffered saline solution without Mg2+ and Ca2+, 20 mM HEPES, 0.1% fatty acid free BSA). Cells were resuspended in assay buffer containing 1X Hanks buffered saline solution without Mg2+ and Ca2+, 20 mM HEPES, 0.1% fatty acid free BSA and 0.5% DMSO. Using online fluidics, 10μl of assay buffer was added to the 384-well microtiter plate and spun at 1000 rpm for 2 minutes to remove air bubbles. 35,000 cells per well were then added in 10μl volume using fluidics. A baseline reading was established for 15 minutes followed by fluidics addition of 10 μl of 3X agonist in assay buffer. MCP- 1 and MIP-1α final assay concentration was 30 nM. Impedance responses were measured for 30 minutes. For pertussis toxintreated samples, THP-1 cells were treated overnight with 100ng/ml of pertussis toxin.
For data analysis, the impedance response due to changes in extracellular current (dZeic) is evaluated. Specifically, the maximum change of the kinetic response from baseline to 10 minutes after stimulation is measured. For dose-response data, the maximum change of kinetic response for each dose of compound is calculated and the curves are fitted to the four-parameter logistic equation.
Additional SAR Assays
The cAMP assay was performed as suggested. Binding and GTPγS assays were performed based on classical radiolabel methods with 125I-labeled ligand and 35S labeled GTPγS [10] . Phospho-ERK was performed as instructed 1/2 assay Kit. β-Arrestin assay was performed based on the β-Arrestin assay direction.
Results and Discussion
Emerging Needs of Labelfree Technology Platforms for GPCR Drug Discovery
Today, about 25% of the top 200 marketed drugs target GPCRs [11-17]. A total of 900 GPCRs have been revealed in the human genome [12, 18, 19]. Therefore, understanding of the molecular functionality of these receptors is highly clinically and commercially relevant. During the past two decades, GPCR technology development played an essential role in GPCR drug discovery from target identification to lead optimization. These advanced technology platforms significantly enriched our knowledge of GPCR pharmacology. For the last few years, it has been evident that a GPCR paradigm shift transitioned the receptor pharmacology from one ligand, one receptor and single efficacy to multiple ligands, differential pharmacology and numerous mechanisms of action (see Figure 2). GPCR drug hunters and thought leaders have started to advocate or accept this paradigm shift. The impact of such evolving concepts can be immense both in terms of screening technology platforms and finding novel mechanisms of GPCR targets. As a result, a single readout using recombinant receptors is no longer meeting GPCR project needs throughout the lifetime of GPCR drug discovery programs. Multiple readouts targeting pyhsiologically relevant systems are often required to discover any differential pharmacology. As a result, cellbased assays have become an important part of the GPCR drug discovery process allowing for investigation of GPCRs and their signaling pathways in a more physiological setting compared to biochemical assays.
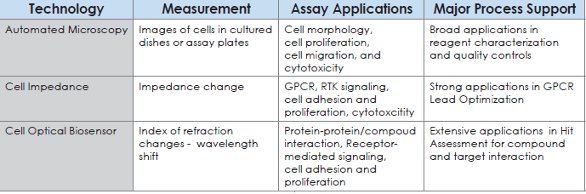
Table 1- Summary of label technology platforms used to support the drug discovery process.
In the past, conventional label technology platforms such as Scintillation Proximity Assay (SPA), FLIPR and cAMP have been broadly implemented to study GPCR/ ligand pharmacology using recombinant systems. One of the main hurdles in the cellbased assay field is to design sufficiently robust assays with adequate signal-to-noise parameters while maintaining the inherent physiology of the pathway or target being investigated. To overcome this issue, the recombinant systems often become the first option to initiate GPCR drug discovery programs. However, measurement of GPCR and ligand binding interactions always requires modification of the binding partners with labels and the recombinant systems for GPCR functional activity demand cellline engineering and over-expression of given receptors. To expand our ability to study GPCR pharmacology, label-free technologies have recently been rapidly developed and became another powerful platform for GPCR drug screening because these label-free technologies preclude the need for cellular labeling or over-expression of reporter proteins. Utilizing non-invasive measurements allowing for time resolution and kinetics, these platforms provide quantitative readouts for various cellular assays through inherent morphological and adhesive characteristics of physiologically relevant cells. In Table 1, we summarize the label technology platforms which could be used to support the drug discovery process into three categories. The first category of platforms is based on automated microscopy. Images of cells in cultured flasks and assay micro-plates are recorded for cell morphology, cell proliferation, cell migration and cytotoxicity. Currently, this platform is broadly implemented to support reagent characterization and cell quality control. The second category is to measure the changes in dielectric impedance of a cell layer that occur in response to the GPCR stimulation, RTK signaling, cell adhesion, cell proliferation and cell cytotoxicity. This platform has demonstrated its strong utility to integrate with other label technology platforms to support GPCR lead optimization. The third platform is to utilize cell optical biosensors through index of refraction changes for monitoring protein/ protein, protein/compound interaction and receptor-mediated signaling. So far, this platform has showed some success in hit assessment for compound and purified protein interaction (data not shown). The application of optical biosensors for GPCR pharmacological studies is limited and will not be discussed in this paper.

Figure 3- Leveraging GPCR technology platforms enabled a paradigm shift in providing high-throughput, comprehensive in-vitro pharmacology data packages.
Enabling Multi-modality Identification for GPCR Drug Discovery Programs
Technology advances in the design of in vitro GPCR assays during the past two decades made possible the integration of disease-relevant biological systems into GPCR drug discovery processes. Although GPCR biochemical ligand-binding assay were the primary screening platform two decades ago, the majority of today’s GPCR drug discovery projects applies functional, cell-based assays for hit identification and lead optimization [20-24].This shift is a result of our increased understanding of the GPCR signaling pathways [25-27] and the desire to use cell-based assays for GPCR positive and negative modulator identification [13, 28, 29]. Figure 3 illustrates that implementation of state-of-the-art and cutting-edge platforms certainly made a significant impact on transition from sole agonist and antagonist identification approaches to multi-modality identification approaches. In 2006, our GPCR pharmacological data packages focused on measurements at the receptor [30], G-protein[31, 32], G-protein dependent and independent pathway levels [15, 21, 22, 33-35] (Figure 3). The results from these platforms were applied for understanding structure activity relationship (SAR) of GPCR agonist and antagonist programs. By 2010, a high-throughput pharmacological data package became available for the GPCR project teams. This expanded capability and allowed GPCR project teams to conduct much more sophisticated pharmacologic characterization including agonism, antagonism, allosteric effects (positive and negative modulators) [28, 36], receptor kinetics and phenotypic responses [23] (Figure 3). This kind of data package can be used not only for SAR but also for target activity relationship (TAR) and structure liability relationship (SLR) (Figure 3). More importantly, the parallel TAR, SAR and SLR significantly enhanced our ability to connect and enrich our knowledge from target ID to lead optimization (Figure 1). Today, high-throughput GPCR comprehensive in-vitro data packages play a critical role on influencing early decisions for GPCR drug discovery programs.
Ensuring High Quality Cell-based Data for GPCR Drug Discovery Programs Through Rigorous Reagent Characterization and Cell Quality Controls
The successful application of cell-based assays for GPCR drug discovery projects requires a detailed understanding of the cellular systems and a clear focus not only on the feasibility and robustness, but also on the disease and biological relevance of the results generated. Clearly, recombinant cell lines used for the GPCR studies have indicated many hurdles to translate conclusions obtained from in vitro assays to in vivo results [27, 30, 37]. There was a palpable trend of introducing primary and native cell lines to support GPCR discovery programs during the past ten years. Selecting appropriate cellular systems becomes an important task and often requires the design of project-specific, rather than generalized and uniform solutions. Regardless of our choices, the quantification of cellular growth, including proliferation and viability must be essential in any laboratory working on cell-based studies. Such techniques enable both the optimization of cell culture conditions and the determination of growth factors.
Traditional methods for cell proliferation and viability measurement can be complicated and time consuming. Labeled reagents and tedious experimental procedures are required to draw a final conclusion [38, 39]. In addition, these molecular labels can disrupt the accurate measurement of kinetic constants and result in antibody cross reactions. Therefore, it is very difficult to implement such technology platforms to support daily cell culture operation. Here, we present an alternative approach for real-time and rapid tracking of cell growth and phenotypic characteristics using automated microscopy.

Figure 4- Real-time tracking of cell growth and phenotypic characteristics using automated microscopy. A) Cell morphology of A875 cells under seeding densities of 300 cells/well, 500 cells/well and 800 cells/well at time 24, 44 and 90 hours. B) Time course of A875 cell confluence under 3 different seeding densities, 300 cells/well (blue), 500 cells/well (pink), and 800 cells/well (green). C) IC50 values of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) cytotoxicity assays at 90 hr under 3 different seeding densities.
Automated microscopy [40, 41] is a useful label-free platform to monitor live cell growth. In addition to recording cell morphology over time, the technology can also calculate cell proliferation rates and cell doubling time. Furthermore, this system can track toxicity effects upon compound treatment. Using the standard cell scratch method, one can measure cell migration over a period of time. Therefore, this labelfree technology not only provides a platform for cellular reagent characterization, but also can be potentially applied for both target validation and lead optimization assays. In Figure 4A, we show an example of real-time tracking of cell morphology and growth using an automated microscope up to 90 hours for A875 melanoma cells that were plated at three different densities including 300 cells/well, 500 cells/well and 800 cells/well, respectively. As shown in Figure 4B, the cells seeded at 500 cells/ well and 800 cells/well reached confluency before the assay measurement time point at 90 hr. In contrast, the cells seeded at 300 cells/well still retained linear growth up to 90 hours. As a consequence of these growth characteristic differences, IC50s of compound 2 and 3 obtained from these three different conditions can shift as much as 10 fold, while compound 1 and 4 under these three different conditions remained similar. These results indicate that cell growth rate should be closely monitored for cellbased assays in order to generate reliable SAR during the lifetime of lead optimization. In summary, using automated microscopy to monitor live cell growth rate is a simple and rapid approach. Otherwise, laborious manual cell counting using dyes would be required at different time points and this technical difficulty prevents the integration of cell growth rate measurements into weekly SAR cell-based assays. The labelfree technology in this study demonstrated its significant advantages.
Leveraging Label-free Technology Platforms to Understand GPCR Signaling Cascades
It is well known that GPCR signaling pathways play an essential role on drug candidate specificity and efficacy [42, 43]. To understand GPCR signaling events, multiple technology platforms such as calcium mobility, cAMP , IP3, β-arrestin, receptor internalization, etc. have been broadly applied to monitor the GPCR signaling cascade at different end points. Conversely, label-free cellular dielectric spectroscopy (CDS) technology[44-47] is a universal platform for the pharmacological evaluation of different classes of GPCRs (i.e., Gs-, Gi-, Gq-coupled GPCRs) with the ability to differentiate between GPCR subtypes without the development of separate assays to monitor secondary messengers associated with G-protein coupling. With CDS technology, it is possible to determine the G-protein coupling of a specific GPCR since differently coupled GPCRs generate different effects on cell morphology that affect impedance measurements. For example, activation of Gs-coupled GPCRs lead to actin depolymerization resulting in a decrease in impedance whereas Gq and Gi coupled GPCRs lead to increased actin polymerization resulting in increased impedance [47].Figure 5 depicts the characterization of an agonist-induced, glucagon-related peptide 1 receptor (GLP-1R) recombinantly expressed in Chinese Hamster Ovary (CHO) cells which displays the general characteristics of a Gs-coupled CDS response after GPCR activation (Figure 5A). Additionally, increased impedance measurements typical of Gi and Gq responses were seen after ligand stimulation of the dopamine D2 short receptor (Figure 5B) and the muscarinic M1 receptor (Figure 5C), respectively. This technology provides an excellent measurement of the GPCR coupling process in cellular systems as it relates to agonistinduced signaling responses.

Figure 5- Cellular Dielectric Spectroscopy used to monitor diverse G-protein coupling signaling pathways. A). Activation of the glucagon-like peptide 1 receptor (GLP1R) , a Gs-coupled receptor, was stimulated by addition of 100 nM GLP-1 in GLP-1 expressing CHO cells. B). The dopamine D2 short receptor (D2sR) in CHO was stimulated by addition of 1 μM dopamine to yield a response profile typical of Gi -coupled GPCRs. C) CHO cells expressing the muscarinic M1 receptors (M1R) were treated with 10 μM carbachol and demonstrated a Gq CDS response profile. D). CDS profiles for a Gq-coupled GPCR after endogenous ligand stimulation in routinely cultured cells (blue line) and cryopreserved cells (red line). E). Comparison of concentration response curves for a Gq-GPCR agonist using routinely cultured cells (blue line) and cryopreserved cells (red line).
One of the challenges to use a broad spectrum of cell-based assays to support GPCR drug discovery processes is resource demands on just-in-time cellular reagents. Cyropreservation represents a significant process improvement for high throughput screening [48, 49] for hit identification, hit assessment and lead optimization. Cryopreserved cells are frozen cells which are thawed at the time of assay performance. Cryopreserved cells have many advantages including the ability to scale, pool, freeze and quality control large batches of cells, decreased time required to perform assay, allows spikes in assay demand by libraries to be easily managed and assay cycle time can be reduced to run more compounds more frequently for decreased data turn-around time. The major impact comes in savings of FTE time and reagent costs as well as more flexibility in assay scheduling. Label-free CDS technology was used to compare CDS profiles of cryopreserved cells and routinely cultured CHO cells expressing a Gqcoupled GPCR (Figure 5D). A consistent CDS response profile (Figure 5D) and agonist concentration response curves (Figure 5E) for both cryopreserved and routinely culture cells indicated the usefulness of the technology for the characterization of cryopreserved reagents in terms of thaw conditions (i.e., at time of assay vs. overnight), G-protein coupling recovery time after thaw and calibrating the effectiveness of agonist-induced signaling after thaw. In conclusion, cryopreserved cells can be implemented to support the label-free CDS technology platform and provide great flexibility to integrate labelfree technology platforms into different stages of drug discovery processes.
Driving GPCR Mechanistic Studies for Deeper Understanding of GPCR Pharmacological Profiles
GPCR cellular responses are mediated through a number of signaling pathways as shown in Figure 6A. For example, Gq and Gi/o signaling pathways trigger increased actin polymerization through the calcium/ PKC pathway or through a reduction in cAMP. Increases in cAMP production reduce actin polymerization mediated by activated Gs-coupled GPCRs. Within the signaling process, there are a number of signaling events that take place to lead to the cellular shape changes that can affect impedance. The CDS technology provides a simple and rapid way to reflect these signaling events. However, to better characterize the CDS profile, it is important to use pharmacological modulators of these pathways for detailed studies on GPCR agonist mechanism of action. Figure 6B lists a number of reagents available to probe Gs-, Gi- and Gq-mediated processes. These tools become especially important when there are no known agonist tool molecules for a given receptor, when characterizing an orphan GPCR, when studying promiscuously coupled GPCRs or when dissecting biased agonism [50]. The data Figure 6C are an example of how to use the CDS technology platform to confirm the signaling mechanisms of chemokine receptors, CCR1 and CCR2. The effects of MIP-1α, the endogenous ligand for chemokine receptor CCR1, and MCP-1, the endogenous ligand for chemokine receptor CCR2, were evaluated in THP-1 human acute monocytic leukemia cells, and changes in impedance were detected in the presence of these agonists (Figure 6C). An overnight pre-treatment with pertussis toxin abolished the impedance response to either CCR1 or CCR2 agonists, thus confirming that MIP-1α and MCP-1 are signaling through Gi-coupled receptors.
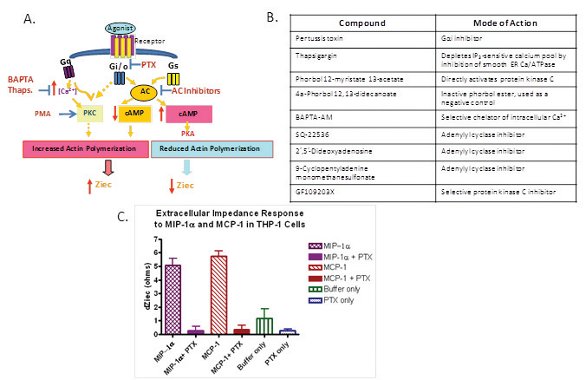
Figure 6- Cell impedance technology platform applied to detailed GPCR mechanistic studies for target validation and lead optimization. A) Diagram of key modulators for GPCR signaling pathways. B) Mode-of-action of known GPCR signaling modulators. C) Agonist-induced THP-1 cellular impedance changes are completely abolished by pertussis toxin treatment.
Understanding Differential SAR Profiles Through Integrated Technology Platforms
Receptor binding and/or second messenger assays such as cAMP levels or calcium flux are common and primary assay platforms for GPCR structural activity relationship (SAR) efforts. However, during the lead optimization process, project teams often encounter one main issue. That is the leading compound series no longer shows a clear SAR trend in the established in vitro binding or second messenger assays. This in turn results in substantial pressure on in-vivo efficacy studies because the in-vivo testing capacity is often limited to a few compounds. Selecting differential pharmacological profiles within diverse leading series is a primary goal of the invitro screening strategy to enable efficient in-vivo proof-of-concept (POC) studies. To address lack of compound differentiation in the primary GPCR in-vitro assays, further signaling readouts such as GTPγS, phospho- ERK and β-arrestin assays are often gradually introduced to the lead optimization process to further drive SAR. However, for each GPCR target, a particular signaling endpoint readout can be biased depending on the physiological relevance of signaling. In this situation, a label-free technology platform such as CDS might provide a unique and powerful solution because it integrates multiple signaling end points and potentially becomes an alternative comprehensive readout for the entire cellular pathway event.

Table 2- A heat map of compound activities in diverse GPCR signaling end points demonstrated the utility of the CDS label-free technology platform for SAR differentiation. Compound EC50 values were indicated in five potency categories, including low nM (green), high nM (light green), low mM (yellow), middle mM (orange) and high mM (red).
Here, we present a case study to indicate how to integrate the label-free technology platform into labeled technology platforms for a GPCR program. Data in Table 2 demonstrates that 14 compounds tested in the primary cAMP assay have a potency range from sub nM to low nM for this Gi- coupled GPCR target. Based on the previous in-vivo results (data not shown), this kind of SAR separation range was not sufficient for compound selection decisions for further in-vivo POC testing. Additional binding and GTPγS assays were then conducted, resulting in a 5-fold increase in compound separation ranges (Table 2). Signaling end points, such as β-arrestin, phospho-ERK and label-free CDS assays were next performed in order to further differentiate these compounds. Indeed, the activities of these 14 compounds were clearly set apart in these last three assays with 5 potency ranges from low nM to high nM, low μM, middle μM and high μM. A compound potency separation fold for a given leading series can be determined by a ratio of the weakest compound over the most potent compound. The heat map in the Table 2 indicates that the compound SAR was dramatically discriminated in the label-free assay with over 3000-fold separation (Table 2). In conclusion, the label-free platform, CDS technology is an assay platform which could combine different signaling readouts and classify the 14 compounds into 5 different categories based on their potency. Further in vivo experiments are ongoing to explore if this integrated approach would be ideal for this target to differentiate compounds in vitro and build good connectivity between in-vitro pharmacology and in-vivo efficacy.
Future Perspectives
In summary, we have demonstrated that two label-free technologies, automated microscopy and cellular dielectric spectroscopy, are very useful technology platforms for reagent evaluation, target tool molecule identification and compound characterization. These platforms can be selectively integrated with other technology platforms to support GPCR drug discovery process from target ID to lead optimization. We predict that these labeled technology platforms will be broadly applied across the entire drug discovery processes and beyond GPCR target classes in the next decade. One can envision that these label-free technology platforms can be broadly applied to monitoring any cellular events which lead to cellular morphology changes and/or mass redistribution. The main advantage of such label-free technology is its ability to capture phenotypic responses in the native context where other labeled technology platforms have shown palpable limitations in these settings. However, random implementation of label-free technology platforms to suppor drug discovery programs sometimes can be destructive because the phenotypic readout isn’t always the best, the most rapid and the most rational approach during drug hunting processes. Depending on target classes and stages of drug discovery processes, label-free technology platforms can play both valueadded and non-value added roles. The key for leveraging label-free technology applications is to selectively integrate these platforms with other technology platforms. A comprehensive data package will permit earlier decisions. The right technology solutions will guide drug discovery programs onto the right path. With future improvement in sensitivity and throughput, we believe that label-free technology platforms will bring irreplaceable impacts on both small and large molecule drug discovery efforts in the future.
Acknowledgments
We appreciate the dedication and experimental work support from Jing Chen, Richard Ryan, Ming Lei, Sha Li, Chi Sum, John Lehrach, Sarah Malmstrom, Rajasree Golla and Ge Zhang.
References
- Lunn, C.A., Label-free screening assays: a strategy for finding better drug candidates. Future Med Chem, 2010. 2(11): p. 1703-16
- Abbas, A., M.J. Linman, and Q. Cheng, New trends in instrumental design for surface plasmon resonance-based biosensors. Biosens Bioelectron, 2011. 26(5): p. 1815-24.
- Rapp, B.E., F.J. Gruhl, and K. Lange, Biosensors with label-free detection designed for diagnostic applications. Anal Bioanal Chem, 2010. 398(6): p. 2403-12.
- Lee, P.H., Label-free optical biosensor: a tool for G protein-coupled receptors pharmacology profiling and inverse agonists identification. J Recept Signal Transduct Res, 2009. 29(3-4): p. 146-53.
- Qavi, A.J., et al., Label-free technologies for quantitative multiparameter biological analysis. Anal Bioanal Chem, 2009. 394(1): p. 121-35.
- Fang, Y., A.G. Frutos, and R. Verklereen, Label-free cell-based assays for GPCR screening. Comb Chem High Throughput Screen, 2008. 11(5): p. 357-69
- Proll, G., et al., Potential of label-free detection in high-content-screening applications. J Chromatogr A, 2007. 1161(1-2): p. 2-8.
- Cooper, M.A., Optical biosensors: where next and how soon? Drug Discov Today, 2006. 11(23-24): p. 1061-7.
- Fang, Y., Label-free cell-based assays with optical biosensors in drug discovery. Assay Drug Dev Technol, 2006. 4(5): p. 583-95.
- Rodgers, G., et al., Development of displacement binding and GTPgammaS scintillation proximity assays for the identification of antagonists of the microopioid receptor. Assay Drug Dev Technol, 2003. 1(5): p. 627-36.
- Overington, J.P., B. Al-Lazikani, and A.L. Hopkins, How many drug targets are there? Nat Rev Drug Discov, 2006. 5(12): p. 993-6.
- Strotmann, R., et al., Evolution of GPCR: change and continuity. Mol Cell Endocrinol, 2011. 331(2): p. 170-8.
- . Smith, N.J., K.A. Bennett, and G. Milligan, When simple agonism is not enough: emerging modalities of GPCR ligands. Mol Cell Endocrinol, 2011. 331(2): p. 241-7.
- Jean-Alphonse, F. and A.C. Hanyaloglu, Regulation of GPCR signal networks via membrane trafficking. Mol Cell Endocrinol, 2011. 331(2): p. 205-14.
- Scott, C.W. and M.F. Peters, Label-free whole-cell assays: expanding the scope of GPCR screening. Drug Discov Today, 2010. 15(17-18): p. 704-16.
- Musnier, A., et al., GPCR signalling to the translation machinery. Cell Signal, 2010. 22(5): p. 707-16.
- Dowal, L. and R. Flaumenhaft, Targeting platelet G-protein coupled receptors (GPCRs): looking beyond conventional GPCR antagonism. Curr Vasc Pharmacol, 2010. 8(2): p. 140-54
- Weill, N., Chemogenomic Approaches for the Exploration of GPCR Space. Curr Top Med Chem, 2011. 11(15): p. 1944-55.
- Crossley, R., J.A. Macritchie, and M.J. Slater, Thematic Analysis: A Chemogenomic Approach to GPCR Drug Discovery. Curr Top Med Chem, 2011. 11(15): p. 1925-43.
- Insel, P., et al., GPCR expression in tissues and cells: Are the optimal receptors being used as drug targets? Br J Pharmacol, 2011
- Ross, D.A., et al., Multiplexed assays by high-content imaging for assessment of GPCR activity. J Biomol Screen, 2008. 13(6): p. 449-55.
- Siehler, S., Cell-based assays in GPCR drug discovery. Biotechnol J, 2008. 3(4): p. 471-83.
- Eglen, R.M., Emerging concepts in GPCR function--the influence of cell phenotype on GPCR pharmacology. Proc West Pharmacol Soc, 2005. 48: p. 31-4.
- Ghosh, R.N., et al., Cell-based, highcontent screen for receptor internalization, recycling and intracellular trafficking. Biotechniques, 2000. 29(1): p. 170-5.
- Re, M., et al., The human gonadotropin releasing hormone type I receptor is a functional intracellular GPCR expressed on the nuclear membrane. PLoS One, 2010. 5(7): p. e11489.
- Swanson, R. and J.R. Beasley, Pathwayspecific, species, and sub-type counterscreening for better GPCR hits in high throughput screening. Curr Pharm Biotechnol, 2010. 11(7): p. 757-63.
- Sjogren, B., L.L. Blazer, and R.R. Neubig, Regulators of G protein signaling proteins as targets for drug discovery. Prog Mol Biol Transl Sci, 2010. 91: p. 81-119.
- Lee, J. and M.E. Jung, 5-HT2C receptor modulators: a patent survey. Expert Opin Ther Pat, 2010. 20(11): p. 1429-55.
- Keov, P., P.M. Sexton, and A. Christopoulos, Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology, 2011. 60(1): p. 24-35.
- van den Burg, E.H. and I.D. Neumann, Bridging the gap between GPCR activation and behaviour: oxytocin and prolactin signalling in the hypothalamus. J Mol Neurosci, 2011. 43(2): p. 200-8.
- Frang, H., et al., Nonradioactive GTP binding assay to monitor activation of g protein-coupled receptors. Assay Drug Dev Technol, 2003. 1(2): p. 275-80.
- Bidlack, J.M. and A.L. Parkhill, Assay of G protein-coupled receptor activation of G proteins in native cell membranes using [35S]GTP gamma S binding. Methods Mol Biol, 2004. 237: p. 135-43.
- Hansen, K.B. and H. Brauner-Osborne, FLIPR assays of intracellular calcium in GPCR drug discovery. Methods Mol Biol, 2009. 552: p. 269-78.
- Thomsen, W., J. Frazer, and D. Unett, Functional assays for screening GPCR targets. Curr Opin Biotechnol, 2005. 16(6): p. 655-65.
- Tilley, D.G., G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ Res, 2011. 109(2): p. 217-30.
- Leach, K., P.M. Sexton, and A. Christopoulos, Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci, 2007. 28(8): p. 382-9.
- Mead, E.J., et al., Identification of the limitations on recombinant gene expression in CHO cell lines with varying luciferase production rates. Biotechnol Bioeng, 2009. 102(6): p. 1593-602.
- Gloeckner, H., T. Jonuleit, and H.D. Lemke, Monitoring of cell viability and cell growth in a hollow-fiber bioreactor by use of the dye Alamar Blue. J Immunol Methods, 2001. 252(1-2): p. 131-8.
- Schmid, I., Assessment of viability, immunofluorescence, and DNA content. Curr Protoc Cytom, 2001. Chapter 7: p. Unit 7 11.
- Milewski, R.J., et al., Automated processing of label-free Raman microscope images of macrophage cells with standardized regression for high-throughput analysis. Immunome Res, 2010. 6: p. 11.
- Moller, C. and M. Slack, Impact of new technologies for cellular screening along the drug value chain. Drug Discov Today, 2010. 15(9-10): p. 384-90.
- DeWire, S.M. and J.D. Violin, Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ Res, 2011. 109(2): p. 205-16.
- van den Driesche, S., et al., A label-free indicator for tumor cells based on the CH2-stretch ratio. Analyst, 2011. 136(11): p. 2397-402.
- Peters, M.F., et al., Comparing label-free biosensors for pharmacological screening with cell-based functional assays. Assay Drug Dev Technol, 2010. 8(2): p. 219-27.
- McGuinness, R.P., et al., Enhanced selectivity screening of GPCR ligands using a label-free cell based assay technology. Comb Chem High Throughput Screen, 2009. 12(8): p. 812-23.
- Peters, M.F., et al., Evaluation of cellular dielectric spectroscopy, a whole-cell, label-free technology for drug discovery on Gi-coupled GPCRs. J Biomol Screen, 2007. 12(3): p. 312-9
- Verdonk, E., et al., Cellular dielectric spectroscopy: a label-free comprehensive platform for functional evaluation of endogenous receptors. Assay Drug Dev Technol, 2006. 4(5): p. 609-19.
- Chen, J., et al., Application of largescale transient transfection to cell-based functional assays for ion channels and GPCRs. Methods Enzymol, 2010. 485: p. 293-309.
- Kamiguchi, N., et al., A 96-well plate assay for CYP4503A induction using cryopreserved human hepatocytes. Drug Metab Dispos, 2010. 38(11): p. 1912-6.
- Peters, M.F. and C.W. Scott, Evaluating cellular impedance assays for detection of GPCR pleiotropic signaling and functional selectivity. J Biomol Screen, 2009. 14(3): p. 246-55.
Author Biographies
Litao Zhang is an Executive Director of Lead Evaluation and Mechanistic Biochemistry at Britstol-Myers Squibb, has accumulated more than 10 years of working experience with major pharmaceutical companies, including DuPont, Aventis and Bristol-Myers Squibb. Since 2003, she has established a centralized lead optimization screening function by leveraging technology platforms. Dr. Zhang received her Ph.D. in Biochemistry from Washington University School of Medicine, St. Louis, and completed her post-doctoral training at the University Pennsylvania.
Liang Schweizer is a Sr. Principal Scientist of Lead Evaluation, Molecular Sciences and Candidate Optimization, Bristol- Myers Squibb, oversees numerous in vitro pharmacology projects across different disease areas. Prior to that, she worked as a senior research investigator in the BMS oncology department, leading cancer drug discovery programs. Liang received her Ph.D. from the University of Zurich in 1999 and her post-doctoral training at Memorial Sloan Kettering Cancer Center.
Mary Ellen Cvijic is a Principal Scientist of Lead Evaluation in Molecular Sciences and Candidate Optimization at Bristol- Myers Squibb. Her team focuses on invitro bioassay design and technology implementation for lead optimization for broad impact across the discovery portfolio. Dr. Cvijic received her Ph.D. in Cellular and Molecular Pharmacology from Rutgers University and The University of Medicine and Dentistry of NJ and her post-doctoral training from The Pennsylvania State University College of Medicine.
This article was printed in the 11/14/2011 issue of International Drug Discovery,6,, 5,. Copyright rests with the publisher.