Abstract
Registration of small molecule pharmaceuticals requires a comprehensive assessment of their genotoxic potential using a standard battery of tests. The genetic toxicity tests of the standard battery are in a period of re-evaluation to assess their utility in human hazard identification and risk assessment. There are concerns with the in vitro mammalian cell assays with respect to the high incidence of positive results and their poor correlation in predicting carcinogenicity. The positive results can necessitate follow-up testing. Approaches for dealing with a positive finding include a weight-of-evidence approach, assessment of mechanism of action, and additional supportive studies.
Introduction
Registration of small molecule pharmaceuticals requires a comprehensive assessment of their genotoxic potential using a standard battery of tests [1, 2]. These tests consist of in vitro and in vivo tests designed to detect compounds that induce genetic damage by various mechanisms. It is clear that no single test is capable of detecting all relevant genotoxic agents [1, 2]. Therefore, the usual approach is to conduct a battery of in vitro and in vivo tests for genotoxicity. Such tests are complementary rather than representing different levels of hierarchy. The general features of a standard test battery as defined by the ICH S2B Guidance [2] can be outlined as follows:
- An in vitro bacterial reverse mutation test
- An in vitro mammalian cell test that detects clastogenic effects and/or gene mutations
- An in vivo test for chromosomal damage in rodents could be either an analysis of chromosomal aberrations in bone marrow cells or an analysis of micronuclei in bone marrow or peripheral blood erythrocytes.
The Center for Drug Evaluation and Research (CDER) generally views the results of the standard battery of genotoxicity tests genotoxicity findings as an early predictor of carcinogenicity for the protection of clinical trial subjects during drug development although it is noted that genetic damage can also be precursors to other diseases besides cancer [3]. Most drugs will undergo carcinogenicity testing prior to drug approval, but results are generally not available for clinical trials. Carcinogenicity study results are generally submitted with a new drug application (NDA). A positive finding in a genotoxicity assay can be the basis for a clinical hold when the trial is in healthy volunteers or minimally ill patients and is almost always the basis for additional studies [3, 4]. The sponsor must demonstrate that despite the positive finding, risks to clinical subjects are minimal [3, 4]. This requires additional testing, usually in animals, and can result in delays in development programs [3, 4].
More than 14 to 15 years have elapsed since the ICH S2A and S2B Guidances were implemented and significant experience has been gained in understanding the performance of these in vitro and in vivo genotoxicity tests of the standard battery. The genetic toxicity tests of the standard battery are in a period of re-evaluation to assess their utility in human hazard identification and risk assessment. Concerns have been raised about the performance of the in vitro mammalian cell assays in terms of generating a high incidence of positive results that have poor correlation to the prediction of carcinogenicity [5-8].
Concerns for the In Vitro Mammalian Cell Genetic Toxicity Tests
Snyder and Green [9] reported for approved drugs with at least one standard genetic toxicity result, from the 1999 Physician’s Desk Reference (PDR) and peer-reviewed published literature, positive responses were most frequently observed for the in vitro cytogenetics test (24.8%) and in vitro mouse lymphoma assay (MLA, 25%) as compared to the in vitro bacterial mutagenesis test (8.3%) and in vivo cytogenetics (11.5%). In an update published in 2009 [10] with information from the 1999 to 2008 PDR, these percentages were essentially unchanged.
In data presented at an ECVAM Workshop [6], the German Federal Institute for Drugs and Medical Devices (BfArM) between 1995 and 2005 reviewed data from 804 in vitro mammalian cell chromosomal aberration studies generated from approximately 600 pharmaceuticals. The frequency of positive results in four different cell types (i.e., human lymphocytes, Chinese hamster ovary cells, V79 cells, and Chinese hamster lung cells) studied for chromosomal aberrations and in the mouse lymphoma assay (detecting gene mutations as well as chromosomal damage) was very similar and averaged approximately 30%.
Matthews et al. [11] conducted a retrospective analysis of standard genetic toxicity tests, reproductive and developmental toxicity studies, and 2-year rodent carcinogenicity bioassays to identify the genetic toxicity and reproductive toxicity endpoints whose results best correlated with the 2-year rodent carcinogenicity bioassay. The in vitro chromosome aberration test and mouse lymphoma assay (two scoring systems) did not correlate with carcinogenicity results. It is noted that the most recent IWGT recommendations [12] for assessing performance of the MLA as well as assay scoring were not available at the time of the Matthew et al. [11] analysis.
Kirkland et al. [7] evaluated how the in vitro genotoxicity tests, Ames test, MLA,and a test for clastogenicity (in vitro micronucleus or chromosomal aberration test) performed, in terms of sensitivity and specificity, in discriminating a large number of rodent carcinogens and noncarcinogens. The sensitivities of the MLA and in vitro chromosomal aberration test were relatively high at 80.8 and 69.6%, respectively, while the Ames test was only 60.3%. In contrast, the specificity of the Ames test was relatively high at >70%. However, the specificity of all three of the in vitro mammalian cell tests was very low (47.6 to 55.1%); a non-carcinogen was just as likely to give a positive result as a carcinogen.
CDER experience with drug-related neoplasms found in carcinogenicity studies with rats and mice has indicated that these tumors may be attributed to secondary effects related to the pharmacological actions of the drugs [8]. Positive genetic toxicity results with these drugs, primarily from the in vitro mammalian cell tests, generally appeared to be unrelated to the tumor findings [8].
Alternative Considerations of the In Vitro Mammalian Cell Assays
Elespuru et al. [13] contended that the in vitro mammalian cell assays were the most sensitive tests of the standard battery. It is preferable to improve the assays and their interpretation, rather than to eliminate them, or alter them to the point that they are no longer useful as sentinels for genotoxic activity. The article had a primary focus on the MLA. The in vitro chromosomal aberration assays are the predominant in vitro mammalian cell system used in drug development although there has recently been a small shift to MLA with the revised scoring system [12]. Gollapudi, Schisler, and Moore [14] have conducted a reevaluation of National Toxicology Program MLA data and contend that the quality of the MLA data was poor and could adversely affect any analyses dependent upon the NTP MLA data.
Relevancy of In Vitro Test Conditions to Assessment of Human Hazard Identification and Risk Assessment
Kirkland et al. [15] postulated several mechanisms that could result in positive results from in vitro genetic toxicity tests that were generally not indicative of human hazard. First, chemicals could exert in vitro genotoxic effects by primary damage to a non-DNA target and there would be a no-effect concentration below which there would be no damage to the cellular target or DNA. Second, chemicals might induce damage by a process that is specific to the in vitro test system. Third, chemicals may induce direct damage to DNA, but only at certain concentrations above a threshold defined, for example, by detoxification or other cellular protective processes such as those that occur at extreme or non-physiological conditions. For each of these situations, the no-effect concentration, process, or threshold is generally not achieved in vivo at therapeutic drug concentrations and the in vitro genotoxicity does not appear to be predictive of an in vivo (or human) hazard. For high concentrations of drug tested in an in vitro setting, these positive results unrelated to primary DNA damage might be characterized as off-target drug activities (e.g., primary damage to a non-DNA target such as interference with metabolism of nucleotides and their precursors, damage to spindle proteins, inhibition of DNA synthesis, or inhibition of topoisomerase). As in vitro drug concentrations increase, the potential for off-target activity becomes greater. Further, these in vitro drug concentrations can be potentially unachievable in the in vivo setting due to toxicity. Mechanistic studies to discern these offtarget activities can often be difficult to perform, the results can often be difficult to interpret from a regulatory standpoint, and can lead to delays or cancellation of drug development.
The use of high drug concentrations in the in vitro genetic toxicity tests that are several orders of magnitude higher than therapeutic concentrations has been an area of particular controversy. The highest recommended concentration for the in vitro mammalian cell tests (from ICH S2A, Section 2.1.2) is 5000 μg/mL or 10 mM (whichever is the lower) for freely soluble, non-toxic compounds (Section 2.1.2.1). Considerations of cytotoxicity (Section 2.1.2.2) and solubility (Section 2.1.2.3) can lower the highest concentration tested. The ICH S2 (R1) Guidance recommends [5] that the highest concentration for the in vitro mammalian cell tests be lowered to 500 μg/mL or 1 mM (whichever is the lower) for freely soluble, non-toxic compounds. Schulz and Schmoldt [16] provided human therapeutic and toxic concentrations of more than 800 drugs. The compilation includes multiple classes of drugs as well as some toxicologically relevant xenobiotics. Fewer than five (0.6%) of the 800 agents had therapeutic blood, plasma, or serum concentration ranges that exceeded 500 μg/mL (Limit dose proposed by the Step 2 ICH S2 (R1) Guidance [5]). The majority of agents had therapeutic concentrations as well as lower end of the toxic concentration range that were several orders of magnitude lower than 500 μg/mL. The data from Schulz and Schmoldt [16] raises further concerns the current limit dose of 5000 μg/mL or 10 mM (whichever is lower) would appear to be non-physiological and/or not achievable in the in vivo setting due to toxicity, which can lead to questions of relevancy.
With consideration of the proposed change for the limit dose for pharmaceuticals to 500 μg/mL or 1 mM for the in vitro mammalian cell tests, Parry et al. [17] conducted an analysis to assess the appropriateness of the 10 mM or 5000 μg/mL top concentration currently recommended for genotoxicity testing of chemicals. Data for 384 chemicals with available in vitro mammalian cell genotoxicity data were used in the assessment although the analysis principally focused on the MLA, in vitro chromosomal aberration assay and Ames assay for which the highest amount of data was available. The assessment determined that 24 (6.25%) compounds with known carcinogenic potential that could not be detected by the commonly used test battery if the maximum testing concentration was lowered from 10 to 1 mM. Kirkland and Fowler [18] conducted assessments of these 24 rodent carcinogens and determined that only one compound, methylolacrylamide, a low molecular weight compound, might require testing at concentrations >1 mM. It is generally recognized that low molecular weight compounds (MW<200) require testing at higher concentrations (i.e., consideration of molar concentrations only could result in testing at inadequate concentrations).
Coping with a Positive Result
A positive finding in a genotoxicity assay will almost always require additional followup studies. The Guidance for Industry and Review Staff entitled “Recommended Approaches to Integration of Genetic Toxicology Study Results finalized in January 2006” recommends three approaches for coping with a positive finding in one of the genetic toxicity tests of the standard battery that include (A) weight-of-evidence approach, (B) mechanism of action, and (C) additional supportive studies [4].
Weight-of-Evidence Approach
A weight of evidence (WOE) approach involves an assessment of all available data for the drug candidate to assess the potential for genotoxic hazard. For hypothetical drug candidates A and B, results of the in vitro bacterial reverse mutation test and in vivo micronucleus test were found to be negative; however, positive results were observed with the in vitro Chinese hamster ovary (CHO) cell chromosomal aberration assay. Table 1 displays the results of in vitro CHO cell chromosomal aberration assays with Drug Candidates A and B. Concentrations tested and the observed cytotoxicity with the two agents were identical. For drug candidate A, a positive response was observed although it occurs only at the high dose and the increase was just outside the range for historical control values for the solvent and cell line employed (0 to 5.0%). The WOE approach could potentially indicate that although a small increase in the frequency of chromosomal aberrations was statistically significant, it lacks biological relevance. Contributing considerations could include (1) the level of cytotoxicity at which the response was seen, and (2) corroborating data from the same or complementary assays. In contrast, for drug candidate B, there were dose-related increased incidences of aberrations that were well outside of the historical control values.
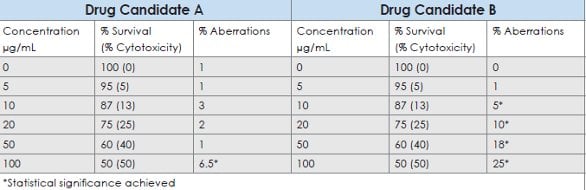
Table 1- Hypothetical results of Chinese hamster ovary cell chromosomal aberration assays with Drug Candidates A and B
Mechanism of Action
Positive results can sometimes be explained by knowledge of the mechanism of action (MOA). It has been demonstrated that in vitro clastogenic effects can result from non-physiologic exposure conditions (e.g., excessively high osmolarity or low pH) that were not relevant to human risk. Certain genotoxic responses are thought to have thresholds below which a hazard does not exist such as agents that induce effects by indirect mechanisms (e.g., interference with metabolism of nucleotides and their precursors, damage to spindle proteins, inhibition of DNA synthesis, or inhibition of topoisomerase). The recommended approach would be presentation of evidence demonstrating a threshold that would not be attained during the proposed clinical exposure or a mechanism that would not be expected to be operative in vivo. Positive responses that are satisfactorily explained by an MOA may allow clinical studies in normal volunteers or in patients to proceed without additional studies.
Additional Supportive Studies
For hypothetical drug B (above), the result from in vitro CHO cell chromosomal aberration study demonstrated a reproducible positive dose-response. Results from in vivo bone marrow cytogenetic studies were frequently negative, even for many compounds with positive in vitro genetic toxicology results that might be attributed to differences between cultured cells and intact animals such as differing in vitro and in vivo metabolism, metabolic inactivation in the intact animal, failure of the parent compound or metabolite to reach the target cell, or an inability to achieve plasma levels in vivo comparable to in vitro concentrations associated with the positive response.
An additional in vivo genetic toxicity test could be useful in clarifying in vitro positive results [1]. Potential in vivo follow-up tests include:
- Flow cytometry assessments from repeat-dose toxicity studies with mice, rats, dogs, and monkeys can be evaluated for micronucleus induction [19-21]
- Peripheral blood lymphocytes from repeat-dose studies in rats or monkeys can be cultured and assessed for chromosome damage in metaphase spreads.
- DNA damage can be assessed in potential target tissues using an in vivo Comet assay [22]. Available data show that cytotoxicity does not generate increases or decreases in DNA migration in the liver Comet Assay [22]. A liver comet assay has been shown to complement an assessment of micronucleus induction in blood or bone marrow. Integration of the in vivo comet and micronucleus tests into general toxicology studies has been demonstrated to be scientifically acceptable [23]. It is noted that the in vivo unscheduled DNA synthesis assay generally has extremely poor sensitivity.
Future Approaches for Follow-up Testing
Ellinger-Ziegelbauer et al. [24] assessed the ability of toxicogenomic analysis of cellular stress responses to distinguish the mechanisms of action of genotoxicants. To evaluate the utility of such a toxicogenomic analysis, gene expression profiles of TK6 cells treated with four model genotoxic agents were evaluated using a targeted high density real-time PCR approach in a multilaboratory project coordinated by the Health and Environmental Sciences Institute Committee on the Application of Genomics in Mechanism-based Risk Assessment. Expression analysis of a relevant gene set was capable of distinguishing compounds that cause DNA adducts or double strand breaks from those that interfere with mitotic spindle function or cause chromosome damage as a consequence of cytotoxicity. The gene expression profiles could potentially provide information relevant to mechanisms of genotoxic damage and differentiating molecular mechanisms of action of genotoxicants. More compounds need to be tested to identify a robust molecular signature.
Conclusions
A battery approach is still reasonable, because no single test is capable of detecting all genotoxic mechanisms relevant in tumorigenesis. The genetic toxicity tests of the standard battery are in a period of re-evaluation to assess their utility in human hazard identification and risk assessment. There are concerns with the in vitro mammalian cell assays with respect to the high incidence of positive results and their poor correlation in predicting carcinogenicity. Further, use of high concentrations in these in vitro assays appear to be non-physiological and/or not achievable in the in vivo setting and increase the possibility of off-target effects leading to false positive results. The positive results can necessitate follow-up testing. Three approaches for coping with a positive finding in one of the genetic toxicity tests of the standard battery include a weight-of-evidence approach, assessment of mechanism of action, and additional supportive studies. In the near future, toxicogenomic analysis of cellular stress responses can provide insight into mechanisms of action of genotoxicants.
Disclaimer
The views expressed in this article are those of the author and do notnecessarily reflect the views of the US FDA.
Acknowledgements
The Author thanks Dr. David Jacobson- Kram for his assistance and advice in the preparation of this article.
References
- Specific Aspects of Regulatory Genotoxicity Tests for Pharmaceuticals, ICH S2A; http://www.fda.gov/downloads/ Drugs/GuidanceComplianceRegulatory Information/Guidances/UCM074925.pdf.
- S2B Genotoxicity: A Standard Battery for Genotoxicity Testing of Pharmaceuticals, ICH S2B; http://www.fda.gov/downloads/ Drugs/GuidanceComplianceRegulatory Information /Guidances/UCM074929.pdf.
- Jacobson-Kram D and Jacobs A (2005) Use of Genotoxicity Data to Support Clinical Trials or Positive Genetox Findings on a Candidate Pharmaceutical or Impurity . . . . Now What? International Journal of Toxicology 24:129–134..
- Guidance for Industry and Review Staff – Recommended Approaches to Integration of Genetic Toxicology Results; http://www.fda.gov/ downloads/Drugs/Guidance ComplianceRegulatoryInformation/ Guidances/ucm079257.pdf.
- Genotoxicity Testing and Data Interpretation for Pharmaceutical Intended for Human Use ICH S2 (R1); http://www.fda.gov/downloads/Drugs/ GuidanceCompliance Regulatory Information/Guidances/UCM074931.pdf.
- Kirkland D, Pfuhler S, Tweats D, Aardema M, Corvi R, Darroudi F et al. (2007) How to reduce false positive results when undertaking in vitro genotoxicity testing and thus avoid unnecessary followup animal tests: Report of an ECVAM Workshop Mutation Research 628: 31-55.
- Kirkland D, Aardema M, Henderson L, Muller L (2005) Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens I. Sensitivity, specificity and relative predictivity. Mutation Research 584: 1-256.
- Jacobs A (2005) Prediction of 2-Year Carcinogenicity Study Results for Pharmaceutical Products: How Are We Doing? Toxicological Sciences 88: 18-23.
- Snyder RD and Green JW (2001) A review of genotoxicity of marketed pharmaceuticals. Mutation Research 488: 151-169.
- Snyder RD (2009) An update on the genotoxicity and carcinogenicity of marketed pharmaceuticals with reference to in silico predictivity. Environmental and Molecular Mutagenesis 50: 435-450.
- Matthews EJ, Kruhlak NL, Cimino MC, Benz RD, Contrera JF (2006) An analysis of genetic toxicity, reproductive and developmental toxicity, and carcinogenicity data: I. Identification of carcinogens using surrogate endpoints. Regulatory Toxicology and Pharmacology 44: 83-96.
- Moore MM, Honma M, Clements J, Bolcsfoldi G, Burlinson B, Cifone M, et al., (2007) Mouse lymphoma thymidine kinase gene mutation assay: meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, recommendations for 24-h treatment. Mutation Research 627:36-40.
- Elespuru RK, Agarwal R, Atrakchi AH, Bigger CA, Heflich RH, Jagannath DR, et al., (2009) Current and future application of genetic toxicity assays: the role and value of in vitro mammalian assays. Toxicological Sciences 109:172-179.
- Gollapudi B, Schisler MR, and Moore MM (2010). Evaluation of Publicly Available Mouse Lymphoma Assay Data Using Currently Accepted Standards to Establish a Curated Database. Society of Toxicology 49th Annual Meeting Abstract #695.
- Kirkland DJ, Aardema M, Banduhn N, Carmichael P, Fautz R, Meunier JR, Pfuhler S (2007) In vitro approaches to develop weight of evidence (WoE) and mode of action (MoA) discussions with positive in vitro genotoxicity results. Mutagenesis 22: 161-175.
- Schulz M and Schmoldt A (2003) Therapeutic and toxic blood concentrations of more than 800 drugs and other xenobiotics. Pharmazie 58, 447-474.
- &Parry JM, Parry E, Phrakonkham P, and Corvi R (2010) Analysis of published data for top concentration considerations in mammalian cell genotoxicity testing. Mutagenesis vol. 25: 531–538.
- Kirkland D and Fowler P (2010) Further analysis of Ames-negative rodent carcinogens that are only genotoxic in mammalian cells in vitro at concentrations exceeding 1 mM, including retesting of compounds of concern. Mutagenesis 25: 539–553, 2010.
- MacGregor JT, Bishop ME, McNamee JP, Hayashi M, Asano N, Wakata A, et al., (2006) Flow cytometric analysis of micronuclei in peripheral blood reticulocytes: II. An efficient method of monitoring chromosomal damage in the rat. Toxicological Sciences 94: 92-107.
- Harper SB, Dertinger SD, Bishop ME, Lynch AM, Lorenzo M, Saylor M, MacGregor JT. (2007) Flow cytometric analysis of micronuclei in peripheral blood reticulocytes III. An efficient method of monitoring chromosomal damage in the beagle dog. Toxicological Sciences 100: 406-414.
- Hotchkiss CE, Bishop ME, Dertinger SD, Slikker W Jr, Moore MM, Macgregor JT. (2008) Flow cytometric analysis of micronuclei in peripheral blood reticulocytes IV: an index of chromosomal damage in the rhesus monkey (Macaca mulatta). Toxicological Sciences 102: 352-358.
- Rothfuss A, Honma M, Czich A, Aardema MJ, Burlinsone B, Galloway S, et al., (2010) Improvement of in vivo genotoxicity assessment: Combination of acute tests and integration into standard toxicity testing. Mutation Research.
- Rothfuss A, O’Donovan M, De Boeck M, Brault D, Czich A, Custer L, et al., (2010) Collaborative study on fifteen compounds in the rat-liver Comet assay integrated into 2- and 4-week repeat-dose studies. Mutation Research 702: 40-69.
- Ellinger-Ziegelbauer H, Fostel JM, Aruga C, Bauer D, Boitier E, Deng S, et al., (2009) Characterization and Interlaboratory Comparison of a Gene Expression Signature for Differentiating Genotoxic Mechanisms. Toxicological Sciences 110(2), 341–352.
Tim Robison earned his Bachelor of Science degree in Biochemistry and Ph.D. in Pharmacology and Toxicology from the University of California at Davis. He trained as a postdoctoral scientist at Children’s Hospital of Los Angeles. Dr. Robison was a Research Assistant Professor at the University of Southern California within the School of Pharmacy and Division of Neonatology and Pulmonology from 1989 to 1996. His research was focused in the area of lung toxicology and he obtained extramural funding from the National Institutes of Health and the American Lung Association. Dr. Robison joined the Center for Drug Evaluation and Research within the FDA in 1996 as a Pharmacology and Toxicology Reviewer. He initially served in the Division of Gastrointestinal and Coagulation Drug Products and later transferred to the Division of Pulmonary and Allergy Products. Dr. Robison’s work has involved the evaluation of safety studies conducted with small molecule drug and protein therapeutic candidates. He serves on several toxicology subspecialty committees within CDER including the Genetic Toxicology Committee. In 2004, Dr. Robison obtained certification from the American Board of Toxicology. In 2005, he was promoted to the senior reviewer level. He has been co-chair of the CDER Genetic Toxicology Committee since 2006 and a member of the ICH S2 EWG since 2008. In 2011, Dr. Robison was promoted to a Pharmacology and Toxicology Team Leader.
This article was printed in the 11/14/2011 issue of International Drug Discovery,6,, 5,. Copyright rests with the publisher.