Abstract
The last decade has seen a continued growth in the use of fragment-based drug discovery (FBDD) approaches in the identification and optimization of drug candidates. This has yielded a pipeline of FBDD-derived assets undergoing clinical evaluation with the recent approval of vemurafenib as the first marketed drug derived from FBDD approaches. The success of FBDD in producing high-quality drug candidates results from marrying structural biology, biophysical characterization and rigorous medicinal chemistry. We discuss the factors important in determining the success of an FBDD approach and also consider those areas where future advancements may increase the impact and success of FBDD approaches even further.
Introduction
With the increased availability of proteinligand structural information, fragmentbased drug discovery (FBDD)[1] has emerged as an important hit identification method for the discovery of high-quality starting points for drug discovery over the last decade. The concept of finding small molecule fragment binders and then refining and growing them using structurebased design represents perhaps one of the most fundamental and elegant synergies of structural biology and medicinal chemistry to design drug candidates. The field reached a new milestone with the recent FDA approval of Zelboraf (vemurafenib),[2,3] considered the first drug derived from FBDD approaches, for forms of melanoma. The utility of FBDD is further underscored by the expanding number of FBDD-derived molecules now undergoing clinical evaluation (Table 1).
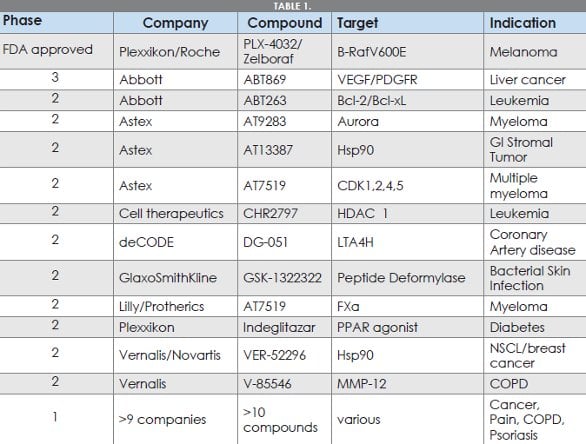
Table 1-Clinical assets discovered through FBDD approaches
FBDD differs from other screeningbased hit identification strategies (i.e., high throughput screening (HTS)[4]) in several ways. First, due to the molecular simplicity of fragments, (typical molecular weight 100-250Da) they more efficiently sample[5-7] a much greater proportion of available chemical space relative to larger HTS screening sets (usually MW in the range 250-500). As a result, FBDD screens can be effective with a fragment screening set comprising as few as 500-1000 well-chosen members and, with suitably sensitive assays, can give ranges of hits containing access to more diversity than an HTS screen comprising a million or more compounds.
Second, due to their small size, fragment hits frequently bind to their targets more efficiently than larger molecules[8]. The importance of efficient binding in allowing more selective, potentially more developable drugs has been recognized and ligand efficiency (ΔG of binding per heavy atom[8-11]) measures are now routinely used in lead optimization. This increased efficiency is likely due to fragments being able to adopt an optimal orientation in the binding site. This optimal orientation may be more difficult for larger molecules to achieve. With careful, structurally guided optimization FBDD hits can deliver ligand efficient clinical candidates which have molecular properties consistent with lower attrition risk through development [12]. In particular, by focusing on optimization of polar interactions, the key parameter of lipophilicity[13] of candidates can be controlled, leading to higher levels of Lipophilic Ligand Efficiency (LLE[12,14]). As more FBDD-derived molecules enter laterstage clinical trials and begin to reach the market, it will be possible to assess whether the structure and property-guided optimization implicit in FBDD is able to translate to producing drugs more efficiently.
Finally, due to their smaller size, the binding affinities of FBDD hits are often much weaker than typically seen with larger, HTSderived hits. This weak affinity necessitates the use of different assay approaches from the functional biochemical readouts most frequently used in HTS approaches. Through use of sensitive biophysical techniques including NMR [15], surface plasmon resonance (SPR) [16], x-ray crystallography [17], thermal denaturation [18], isothermal titration calorimetry[19] and others, protein-ligand interactions can be characterized in an exquisite level of detail. These techniques enable the identification of very weak ligands (most fragment hits typically bind with Kd values in the range 5mM-10μM) but are agnostic to the location of the binding site. Therefore biophysical techniques have the potential to identify hits that interact with allosteric or cryptic binding sites. Many biochemical assays rely on competition with specific binders and therefore may not be able to identify these unique binding interactions. Biophysical techniques also give additional information such as binding stoichiometry, kinetics and identification of confounding assay artefacts (e.g., aggregation-based binding or irreversibility[20]) which often affect biochemical screens but frequently go unnoticed until further into the optimization process.
Elements of a Successful FBDD Campaign
As a result of the advantages of the FBDD approach and the increasing availability of high throughput x-ray crystallography and biophysical assays, many groups are now undertaking FBDD approaches either alone or to complement HTS or other hit generation approaches. FBDD concepts can be successfully applied in a variety of ways with individual strategies often based around particular strengths of specific organizations, but a series of common, fundamental themes emerge across successful FBDD efforts: i) a highquality library of fragments; ii) ready access to protein-ligand structural information; iii) reliable assays to characterize and monitor weak binding and iv) rigorous, structurebased optimization. A typical progression of a FBDD hit identification effort is shown schematically in Figure 1. Effective fragment screening sets can be numerically small in size (e.g., 500-10,000 members) but must be well designed and chemically diverse (for reviews see e.g.,[21] and ref cit) to allow efficient hit followup. Most fragment sets broadly adhere to the rule of three[22] (mw<300, cLogP<3,hydrogen bond acceptors <6 and donors <3) though many focus on smaller molecules allowing even more efficient coverage of diversity space. Due to the high compound concentrations used in FBDD screening,high predicted or measured aqueous solubility (>0.1-2mM) is required. Additional factors such as increased degree of three-dimensionality or the inclusion of “privileged” sub-structures contained within known drugs are also used. Rigorous quality control to remove undesirable molecules (aggregators, redox-labile or reactive molecules) can remove false positives. Removal of these problematic compounds
increases the probability that initial hits will be specific and optimizable binders, which in turn should allow translation of hits to viable lead series more reliably. Whilst an unbiased, structurally diverse fragment set will usually provide good hit rates against a range of targets [23], use of specific sets, designed against particular families of protein targets have also been used to good effect [24].

Figure 1-FBDD-enabled discovery of vemurafenib
Fragment screening is usually performed using a biophysical technique (X-ray, SPR, NMR, or Tm) or robust high-concentration biochemical assay. The assay used must be able to reliably detect hits with weak binding affinities (e.g., ~1mM) and should also be suitable for both identification of initial hits and also for rank ordering analogs (via Kd or IC50 determinations) to support the initial fragment optimization process. Frequently, hits showing activity across multiple assays are prioritized and subsequently have shown higher success rates in x-ray crystallography. If the assay throughput of the primary screen is limited, virtual screening of fragment libraries is also frequently reported. However, docking and scoring of simple fragments may be significantly more challenging than for larger molecules, especially for less efficient binders [25]. Additionally, as small changes to fragments can alter their crystallographically determined binding mode, virtual screening of fragments should be treated with caution.
Whilst it is possible to use crystallography as a primary screen [17]. a more common approach is the combination of biophysical and biochemical assays to triage hits prior to structural determination. As fragment screens frequently give more hits than can reasonably be advanced, a key decision is which of the fragments should be prioritized for optimization with medicinal chemistry. This point is perhaps one of the most crucial as significant chemistry resource is often required for optimization to produce a molecule with appropriate potency and properties to be a useful drug or tool molecule. Factors considered in the prioritization process can include ligand efficiency (and LLE), intellectual property landscape, novelty of binding mode, selectivity against related targets, synthetic tractability and diversity of fragment growth directions. Fragment to lead optimization typically aims to maintain (or increase) efficiency measures (LE & LLE) by making use of structure-based hypotheses to efficiently access novel interactions while increasing compound affinity. While maintaining LE/ LLE will provide highly efficient leads with controlled cLogP, it will not ensure leads with good drug-like properties ( i.e., good solubility, good in vivo pharmacokinetics, appropriate selectivity and lack of problematic CYP450 interactions, etc). The importance of rigorous medicinal chemistry in fragment to lead optimization cannot be understated. In early fragment optimization, binding is frequently weak which requires both routine access to a high-sensitivity assay to drive structure-activity relationships (SAR) and also a team of chemists who are comfortable operating outside of the submicromolar potency ranges usually seen in traditional drug discovery efforts.
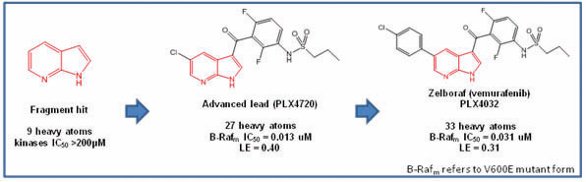
Figure 2-Key parts of FBDD hit identification and progression
To exemplify this process, the discovery of verumafenib[2,3] is summarized in Figure 2. Starting from a weak, non-selective hit (7-azaindole), x-ray crystallographic data (initially in a surrogate kinase Pim-1) allowed optimization to the ligand efficient lead PLX4720. Final optimization to improve the overall profile of the molecule led to PLX4032 (venurafenib) with slightly reduced but still good ligand efficiency. The initial fragment is highlighted in red throughout.
FBDD has found success across many biological targets with the increase in x-ray structural enablement driving the expanding utility [26]. Initial areas of success included kinases [27-31], other ATPases (Hsp90 has been an important vehicle used to test FBDD strategies[32-36]) and others. More recently, expansion to a wider range of soluble targets [37], proteinprotein interaction interfaces [38] and now membrane-bound targets [39,40] has been reported.
A principal attraction of FBDD is the potential low barrier to entry in that large screening sets (cf HTS) are not required. With a wellchosen set of fragments (many are offered commercially) and an affinity/activity assay, a set of hits can be readily generated. Access to x-ray structural determination will also confirm their binding mode but to take full advantage of these fragment hits, they must be optimized to a level of potency (usually 0.1 – 1μM) where they may reliably start to show effects in biological systems. A further significant benefit of FBDD is that this optimization process, because it uses simple starting points, allows maximum flexibility in growing the fragment whilst still keeping the properties (e.g., LogP, mw) in optimal druglike space.
At its heart, FBDD is simply a refined form of high-sensitivity screening/structuredriven medicinal chemistry and can be used alone or in combination with other hit finding approaches (e.g., HTS or rational design). Frequently, integration of FBDD and HTS hit sets can identify overlap to suggest productive optimization avenues increasing the value of the whole data set over that of each screen alone. This integration has found particular use in pharmaceutical companies where the capability to generate HTS and FBDD datasets in parallel together with the ready availability of large corporate and commercial compound collections (frequently >>1M compounds) can be used to rapidly expand structureactivity relationship (SAR) around fragment hits and find more potent, yet still efficient binders. FBDD concepts may also be applied to analyzing HTS datasets in the absence of a direct primary fragment screen: By use of high sensitivity assays on molecules representing substructures of HTS hits, the minimum pharmacophore can be identified prior to regrowing of this derived fragment back to more potent analogs.
Current Issues and Future Directions
Much FBDD work has relied on extensive x-ray structural assistance to guide optimization, but there are targets where x-ray enablement is still not routine and other approaches (e.g., NMR structure) are impractical and hinder an FBDD approach. For example, many protein targets exist as part of multi-component complexes which are frequently unsuitable for x-ray structural determination. Also, in some cases, the isolated protein best suited to crystallography may have limited relevance to the in vivo physiological environment. This leads to optimization guided by systems of limited biological relevance, though this issue is not specific to FBDD approaches of course. Further advancements in the ability to generate structural information using x-ray or other techniques (especially in native or solution phases) will greatly facilitate the optimization process for FBDD and other rational design approaches. The expanded use of FBDD approaches in the absence of structural data[41] may also allow some of these less structurally tractable targets to be tackled. In this regard, further improvements to informatic approaches to rationally guide optimization and SAR studies, especially where they can rely on large corporate collections of readily available analogs, will expand the utility of FBDD.
Given the reliance that FBDD approaches have on translating structural information into high-potency molecules (with good drug-like properties), there is still much to be gained in increasing our understanding of ligand-protein interactions to assist this process. Identifying fragment hits is generally possible with most targets, but reliably translating these hits into drug candidate quality molecules is still not routine and many fragment hits prove not to be optimizable. A better ability to predict what makes some fragments optimizable and others less so would greatly help groups focus on the best series and molecules. As drug discovery begins to tackle new targets, many of them are considered potentially less ligandable based on the topology of their binding sites. There has been a particular focus on modulation of proteinprotein interactions and speculation that interaction over large surface areas may be required for efficient disruption. There is a risk that the small size of fragments may prevent effective interaction with these surfaces but fragment screening for protein-protein interaction inhibitors may still be a powerful approach [42].
Conclusions
Building on the success of generating its first marketed drug, FBDD continues to be a valuable hit identification tool to be used alone or as part of a wider, integrated strategy. If suitable screening sets, assays, structural support and optimization strategies are in place, FBDD is a powerful approach for finding quality hits for a wide range of biological targets. Whilst it is not a panacea and cannot increase the drugability of a poorly tractable or illchosen target, it will usually give the best chance of finding viable chemical matter for interacting with a protein target. The task of optimizing this hit to a molecule with utility as a drug or a tool may be difficult in some cases, but as our ability to generate biologically relevant protein structure and better understand structure-led design increases, the power of FBDD is set to increase further.
References
- R. Carr, A. E., M. Congreve, C. W. Murray, D. C. Rees, Drug Discov Today 10[14], 987-992, 2005.
- J. Tsai, J. T. Lee, W. Wang, J. Zhang, H. Cho, S. Mamo, R. Bremer, S. Gillette, J. Kong, N. K., Haass, K. Sproesser, L. Li, K. S. M. Smalley, D. Fong, Y. L. Zhu, A. Marimuthu, H. Nguyen, B. Lam, J. Liu, I. Cheung, J. Rice, Y. Suzuki, C. Luu, C. Settachatgul, R. Shellooe, J. Cantwell, S. H. Kim, J. Schlessinger, K. Y. Zhang, B. L. West, B. Powell, G. Habets, C. Zhang, P. Ibrahim, P. Hirth, D. R. Artis, M. Herlyn, G. Bollag. Proc. Natl. Acad. Sci. U.S.A. 105[8], 3041-3046. 2008.
- G. Bollag, P. Hirth, J. Tsai, J. Zhang, P. Ibrahim, H. Cho, W. Spevak, C. Zhang, Y. Zhang, G. Habets, E. Burton, B. Wong, G. Tsang, B. West, B. Powell, R. Shellooe, A. Marimuthu, H. Nguyen, K. Y. J., Zhang, D. R. Artis, J. Schlessinger, F. Su, B. Higgins, R. Iyer, K. D’Andrea, A. Koehler, M. Stumm, P.S. Lin, R. J. Lee, J. Grippo, I. Puzanov, K.B. Kim, A. Ribas, G. McArthur, J.A. Sosman, P.B. Chapman, K.T. Flaherty, X. Xu, K.L. Nathanson, K. Nolop. Nature (London, U.K.467[7315], 596-599. 2010.
- R. Macarron, M.N. Banks, D. Bojanic, D.J. Burns, D. A. Cirovic, T. Garyantes, D.V.S. Green, R.P. Hertzberg, W.P. Janzen, J.W. Paslay, U. Schopfer, G. S. Sittampalam. Nat. Rev. Drug Discovery 10[3], 188-195, 2011.
- Hann, M. M., Leach, A. R., Burrows, J. N., and Griffen, E. Compr.Med.Chem.II 4, 435-458. 2006.
- Leach, Andrew R. and Hann, Michael M. Curr.Opin.Chem.Biol. 15[4], 489-496. 2011.
- Blum, Lorenz C. and Reymond, Jean Louis. J.Am.Chem.Soc. 131[25], 8732-8733. 2009.
- Reynolds, Charles H., Tounge, Brett A., and Bembenek, Scott D. J.Med.Chem. 51[8], 2432-2438. 2008.
- Hopkins, Andrew L., Groom, Colin R., and Alex, Alexander. Drug Discov Today 9[10], 430-431. 2004.
- bad-Zapatero, Cele and Blasi, Daniel. Mol.Inf. 30[2-3], 122-132. 2011.
- S. Schultes, C. De Graaf, E.E.J. Haaksma, I.J.P. de Esch, R. Leurs, O. Kraemer. Drug Discovery Today: Technol. 7[3], E157-E162. 2010.
- Leeson, Paul D. and Springthorpe, Brian. Nat. Rev.Drug Discovery 6[11], 881-890. 2007.
- Waring, Michael J. Expert Opin.Drug Discovery 5[3], 235-248. 2010.
- Mortenson, Paul N. and Murray, Christopher W. J.Comput.-Aided Mol.Des. 25[7], 663-667. 2011.
- Lepre, Christopher A. Methods Enzymol. 493[Fragment-Based Drug Design], 219-239. 2011.
- Giannetti, Anthony M. Methods Enzymol. 493[Fragment-Based Drug Design], 169-218. 2011.
- Spurlino, John C. Methods Enzymol. 493[Fragment-Based Drug Design], 321-356. 2011.
- Kranz, James K. and Schalk-Hihi, Celine. Methods Enzymol. 493[Fragment-Based Drug Design], 277-298. 2011.
- Ladbury, John E., Klebe, Gerhard, and Freire, Ernesto. Nat.Rev.Drug Discovery 9[1], 23-27. 2010.
- Giannetti, Anthony M., Koch, Bruce D., and Browner, Michelle F. J.Med.Chem. 51[3], 574-580. 2008.
- Tounge, Brett A. and Parker, Michael H. Methods Enzymol 493, 3-20. 2011.
- Congreve, Miles, Carr, Robin, Murray, Chris, and Jhoti, Harren. Drug Discov Today 8[19], 876-877. 2003.
- Chen, I. Jen and Hubbard, Roderick E. J.Comput.-Aided Mol.Des. 23[8], 603-620. 2009.
- Bamborough, Paul, Brown, Murray J.,Christopher, John A., Chung, Chun wa, and Mellor, Geoff W. J.Med.Chem. 54[14], 5131-5143. 2011.
- vdonk, Marcel L., Giangreco, Ilenia, Hall, Richard J., Korb, Oliver, Mortenson, Paul N., and Murray, Christopher W. J.Med.Chem. 54[15], 5422-5431. 2011.
- Murray, Christopher W. and Blundell, Tom L. Curr.Opin.Struct.Biol. 20[4], 497-507. 2010.
- S. Antonysamy, G. Hirst, F. Park, P. Sprengeler, F. Stappenbeck, R. Steensma, M. Wilson, and M. Wong. Bioorg.Med.Chem.Left. 19[1]. 279-282. 2009.
- S. Howard, V. Berdini, J.A. Boulstridge, M.G. Carr, D.M. Cross, J. Curry, L. Devine, T.R. Early, L. Fazal, A.L. Gill, M. Heathcote, S. Maman, J.E. Matthews, R.L. McMenamin, E.F. Navarro, M.A. O’Brien, M. O’Reilly, D. Rees, M. Reule, D. Tisi, G. Williams, M. Vinkovic, and P. Wyatt. J.Med.Chem. 52[2], 379-388. 2009.
- T.P. Matthews, S. Klair, S. Burns, K. Boxall, M. Cherry, M. Fisher, I.M. Westwood, M.I. Walton, T. McHardy, K.M. Cheung, R. Van Montfort, D. Williams, W.G. Aherne, M.D. Garrett, J. Reader, and I.Collins. J.Med.Chem. 52[15]. 4810-4819. 2009.
- G. Saxty, S.J. Woodhead, V. Berdini, T. G., Davies, M.L. Verdonk, P.G. Wyatt, R.G. Boyle, D. Barford, R. Downham, M.D. Garrett, and R.A. Carr, J.Med.Chem. 50[10], 2293-2296. 2007.
- P.G. Wyatt, A.J. Woodhead, V. Berdini, J.A. Boulstridge, M.G. Carr, D.M. Cross, D. Davis, L.A. Devine, T. Early, R.E. Feltell, J.E. Lewis, R.L., McMenamin, E.F. Navarro, M.A. O’Brien, M. O’Reilly, M. Reule, G. Saxty, L.C. Seavers, D.M. Smith, M.S. Squires, G. Trewartha, M.T. Walker, and A.J.A. Woolford. J.Med.Chem. 51[16], 4986-4999. 2008.
- J.J. Barker, O. Barker, R. Boggio, V. Chauhan, R.K.Y. Cheng, V. Corden, S.M. Courtney, N. Edwards, V.M. Falque, F. Fusar, M. Gardiner, E.M.N. Hamelin, T. Hesterkamp, O. Ichihara, R.S. Jones, O. Mather, C. Mercurio, S. Minucci, C.A.G.N. Montalbetti, A. Muller, D. Patel, B.G. Phillips, M. Varasi, M. Whittaker, D. Winkler and C.J. Yarnold. ChemMedChem 4[6], 963-966. 2009.
- J.R. Huth, C. Park, A.M. Petros, A.R. Kunzer, M.D. Wendt, X. Wang, C.L. Lynch, J. C. Mack, K.M. Swift, R.A. Judge, J. Chen, P.L. Richardson, S. Jin, S.K. Tahir, E.D. Matayoshi, S.A. Dorwin, U.S. Ladror, J.M. Severin, K.A. Walter, D.M. Bartley, S.W. Fesik, S.W. Elmore and P.J. Hajduk. J.Chem.Biol.Drug Des, 70[1], 1-12. 2007.
- C.W. Murray, M.G. Carr, O. Callaghan, G. Chesari, M. “Congreve, S. Cowan, J.E. Coyle, R. Downham, E. Figueroa, M. Frederickson, B. Graham, R. McMenamin, A.M. O’Brien, S. Patel, T.R. Phillips, G. Williams, A.J. Woodhead, and A.J.A. Woolford. J.Med. Chem. 53[16], 5942-5955. 2010.
- Roughley, Stephen D. and Hubbard, Roderick E. J.Med.Chem. 54[12], 3989-4005. 2011.
- A.J. Woodhead, H. Angove, M.G. Carr, G. Chessari, M. Congreve, J.E. Coyle, J. Cosme, B. Graham, P.J. Day, R. Downham, L. Fazal, R. Fettell, E. Figueroa, M. Frederickson, J. Lewis, R. McMenamin, C.W. Murray, A.M. O’Brien, L. Parra, S. Patel, T. Phillips, D.C. Rees, S. Rich, D. M. Smith, G. Trewartha, M. Vinkovic, B. Williams, and A.J.A. Woolford. J.Med.Chem. 53[16], 5956-5969. 2010.
- . S. Johnson, E. Barile, B. Farina, A. Purves, J. Wei, L.H. Chen, S. Shiryaev, Z. Zhang, I. Rodionova, A. Agrawal, S.M. Cohen, A. Osterman, A. Strongin, and M. Pellecchia. Chem.Biol.Drug Des. 78[2]. 211-223. 2011.
- Wilson, C. G. M. and Arkin, M. R. Curr.Top. Microbiol.Immunol. 348[Small-Molecule Inhibitors of Protein-Protein Interactions], 25-59. 2011.
- Congreve, Miles, Rich, Rebecca L., Myszka, David G., Figaroa, Francis, Siegal, Gregg, and Marshall, Fiona H. Methods Enzymol. 493[Fragment-Based Drug Design], 115-136. 2011.
- Congreve, Miles, Langmead, Christopher J., Mason, Jonathan S., and Marshall, Fiona H. J.Med.Chem. 54[13], 4283-4311. 2011. 4
- P. Ray, J. Wright, J. Adam, J. Bennett, S. Boucharnes, D. Black, A. Cook, A.R. Brown, O. Epemolu, D. Fletcher, A. Haunso, M. Huggett, P. Jones, S. Laats, A. Lyons, J. Mestres, J. De Man, R. Morphy, Z. Rankovic, B. Sherborne, L. Sherry, N. Van Straten, P. Westwood, G.Z. R. Zaman. Med.Chem.Lett. 21[1]/ 97-101. 2011.
- C.M. Park, M. Bruncko, J. Adickes, J. Bauch, H. Ding, A. Kunzer, K.C. Marsh, P. Nimmer, A.R. Shoemaker, X. Song, S.K. Tahir, C. Tse, X. Wang, M.D. Wendt, X. Yang, H. Zhang, S.W. Fesik, S.H. Rosenberg, S.W. Elmore. J.Med. Chem. 51[21]. 6902-6915. 2008.
Terry Hughes currently leads a fragment to lead chemistry group at GlaxoSmithKline at their Collegeville site in Pennsylvania, USA. He has over a decade of experience as a medicinal chemist in the pharmaceutical industry working in hit identification through to lead optimization with emphasis on structure-based design. terry.v.hughes@gsk.com
Ian Baldwin has worked for GSK and former companies for 11 years. He has expertise in analyzing data and leading chemistry on many lead discovery programs following HTS campaigns and focused screening. He currently leads a team of fragment based-drug discovery chemists within GSK.
Ian Churcher leads an early stage medicinal chemistry department with responsibility for novel hit identification strategies with global GSK R&D. As well as contributing to the chemical strategy of evolving the GSK corporate screening collection, the group also provides medicinal chemistry support for a portfolio of FBDD programs from hit identification through to lead optimization.
This article was printed in the 11/14/2011 issue of International Drug Discovery,6,, 5,. Copyright rests with the publisher.