Abstract
This article presents a brief overview of both existing and new, emerging strategies for RNA-mediated gene silencing that can potentially be applied as highly selective approaches against abnormal gene expression. Despite the availability of numerous approaches for controlled translational arrest of disease-relevant proteins, none has gained superiority thus far. Therefore, the purpose of this review is to highlight some new strategic directions, which may inspire development of novel therapeutic approaches and thus allow expanding the therapeutic window for drug discovery. Our particular interest is focused on peptidyl-oligonucleotide chemical ribonucleases, although some other approaches for sequence-specific RNA targeting are also presented.
Novel Therapeutic Strategies Based on Gene Silencing
One of the major pharmaceutical challenges today is the development of more powerful, highly selective therapies against abnormal gene expression in disease states, which would allow lower chemotherapeutic doses and thus more tolerable side effects.
Novel therapeutic strategies to selective inhibition of disease states can be facilitated by targeting of upstream components, which are involved in disease initiation (e.g., DNA or messenger RNA) rather than downstream biological pathways. DNA and RNA-mediated gene silencing is recognized as a promising alternative to the conventional approaches, which were traditionally based on treatment of physiological abnormalities at the level of expressed proteins. Controlled translational arrest of certain proteins can be achieved, for example, by specific targeting of mRNA sequences abnormally expressed in disease states (e.g., cancer) leading to a desired therapeutic response [1-8]. Advances in this area might be particularly beneficial for those targets that are not amenable to small-molecule drugs or antibody inhibition [2] (e.g., transcription factors) due to their intracellular location and/or the lack of suitable binding sites for small molecules. Selective targeting of specific genes or messenger RNA sequences [1-8] encoding functional proteins, which are over-expressed in disease states, may potentially expand the therapeutic window for drug discovery.
Gene therapy and antisense oligonucleotide-based approaches demonstrated encouraging results from specific targeting of mRNA abnormally expressed in diseased cells or oncogenes involved in cancer progression [2-8]. Indeed, recent publications and clinical trials have demonstrated the ability of this class of therapeutic agents to significantly suppress expression of target genes via either gene therapy [3, 6] or by targeting specific messenger RNAs using antisense oligonucleotides [1,2, 4,5].
RNA as a Potential Therapeutic Target
An advantage of using RNA as a potential therapeutic target [1, 9] was recognized due to its major role in macromolecular processes (e.g., transcriptional and translational regulation, RNA stability control, RNA splicing and retroviral replication [9]).
Moreover, since RNA is the genetic material of some viruses, the viral genome could possibly be selectively attacked in antiviral chemotherapy. Pathogenic diseases of viral origin represent a global health and socioeconomic problem worldwide. Highly pathogenic RNA viruses include, for example, aggressive strains of corona viruses (SA RS), avian influenza, tick-borne encephalitis and Dengue virus [10, 11]. In addition, prevalence of chronic viral infections such as Human Immunodeficiency Virus (HIV), Hepatitis B virus (HBV) and Hepatitis C virus (HCV) has recently increased [12-14] and can potentially have a significant socioeconomic impact worldwide. Antiviral chemotherapeutics recently approved for treating infections via inhibiting viral propagation are not fully specific and may cause serious side effects and toxicity, especially on long-term treatment. Therefore, there is an unmet medical need for the development of a specific targeting of viral genomic RNA to achieve selective antiviral chemotherapy.
A large number of non-protein-coding RNAs (ncRNA), including micro RNA (miRNA) and short-interfering RNA (siRNA), have recently been identified that have several fundamental functions essential for cell growth, survival and development. This large variety of novel non-protein-coding RNAs have been demonstrated to be involved in gene silencing via RNA-mediated feedback and to play essential roles in controlling all steps of gene expression, including transcription, chromatin modification, epigenetic memory, and alternative splicing. Since the dysregulation of non-protein-coding small regulatory RNAs (e.g., miRNAs) are frequently associated with the development and progression of diseases, particularly genetic disorders, tumour progression, autoimmune diseases and neurodegenerative disease pathogenesis [15], these ncRNA may provide a promising new (miRNA antagonizing) pharmaceutical target [16].
Other advantages of using RNA as a therapeutic target include the lack of repair mechanisms, accessibility of many RNAs to the cytoplasm and a less intensive puckering structure with greater tertiary structural diversity than that found for DNA [17], providing an added potential to target-specific folding motifs as an additional approach to site-specific targeting.
Limitations of the Existing RNA-mediated Gene Silencing Approaches
Although antisense oligonucleotides and gene silencing strategies have demonstrated impressive therapeutic effects [2-8], there are a number of limitations of these approaches, some of which are shared with small drug molecules. These include, for example, the necessity for stoichiometric (1:1) binding between pharmaceutical agent and target, adverse drug reactions, and the absence of catalytic amplification.
There are some other limitations in use of antisense oligonucleotides and their analogues in existing RNA-induced gene silencing approaches. For example, the antisense RNA interference competes with RNA being produced by the cell, and thus the resulting effect is gene knockdown rather than gene knockout [18].
Furthermore, conventional approaches still rely on the paradigm ‘higher affinity translates into higher gene repressing activity’ [4], implying the necessity of using relatively long oligonucleotides (at least 17-20 nucleotide residues) for antisense oligonucleotide-based approaches to ensure specific targeting of a particular stretch of the human genome (or corresponding mRNA sequences). Shorter nucleotide sequences might occur more than once in the human genome and thus shorter antisense oligonucleotides may hybridize at non-targeted regions. On the other hand, use of relatively long antisense nucleotides (or their analogues) may cause non-specific binding due to the ability of longer oligonucleotides to form relatively stable non-perfect hybridization regions with DNA or RNA sequences leading to undesirable off-target effects [19, 20]. Therefore, it is still challenging to strike an appropriate balance to ensure that a unique segment of the human genome is covered whilst eliminating all non-perfect hybridizations.
If the target protein is only over-expressed in the disease state then antisense interference could be highly selective. However, the specificity of RNA targeting might be compromised if relevant RNA is expressed in both normal and malignant cells, which may cause both types of cell to undergo apoptosis leading to undesirable cytotoxicity [21].
Ribonucleases have been seen as an attractive alternative to the conventional gene silencing approaches, and the nonspecific ribonuclease moieties of bacterial or plant protein toxins (e.g., diphtheria toxin or Pseudomonas exotoxin) has been selectively delivered to tumor cells using antibodies or tumor-targeting ligands [22, 23]. Although the results seem to be encouraging, the cytotoxic specificity of these immunotoxin ribonucleases relies wholly on the targeting ligand to prevent wide-scale systemic toxicity [22, 23]. Furthermore, the toxin ribonucleases damage RNA uncontrollably and their high imunogenicity leads to a rapid clearance from the bloodstream [22, 23].
Alternative strategies for eliminating pathogenic gene expression involve sequencespecific, post-transcriptional gene silencing using the RNA interference (RNAi) and related pathways (e.g., microRNA and short-interfering RNAs) [24-27]. Highly potent RNAi offers enhanced therapeutic potential via sequence-specific translational arrest and also benefits from an amplification cascade through the cleavage of perfectly matched RNA targets. However, the biological cascade triggered by siRNAs is complex and may be associated with offtarget effects and toxicity due to saturation of the endogenous RNAi functions [27], raising concerns of their potential safe use in humans [24-26]. The limited duration of gene silencing [27] and problems associated with cellular delivery represent additional issues associated with the siRNAbased approaches.
Sequence-specific Chemical Ribonucleases
The development of novel biocatalytic structures mimicking the active center of natural ribonucleases and capable of cleaving RNA targets can provide a basis for generating new useful biological tools, perhaps even therapeutics, affecting specific messenger RNAs and viral genomic RNAs. Cytotoxic chemical ribonucleases can potentially be applied as a highly selective therapy
against abnormal gene expression thus offering a gene-specific approach to chemotherapy where a disease that involves the production of a harmful protein could be treated by antisense interference with a disease-relevant mRNA.
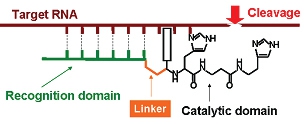
Figure 1: Schematic illustration of a sequence-specific chemical ribonuclease showing its hybridization with the target sequence of RNA via the binding domain.
Chemical ribonucleases (see Figure 1 for schematic illustration) are synthetic supramolecular structures mimicking the active center of natural ribonucleases (Figure 2), which are capable of interacting with RNA molecules and catalyzing cleavage of phosphodiester bonds in RNA sequences.
Sequence-specific artificial ribonucleases can be directed to a specific nucleotide sequence and to specific RNA molecule by conjugating the RNA cleaving catalytic groups (Figure 2) to antisense oligonucleotides capable of recognizing specific regions within the target RNA sequences. These artificial mimics offer the prospective of sequence-specific cleavage of target RNA sequences, which is a major advantage over natural ribonucleases or non-specific ribonuclease moieties of bacterial or plant toxins.
Figure 2: Amino acids, which normally form catalytic centers of natural ribonucleases and used as a scaffold to mimic cleving constructs of chemical ribonucleases.
Sequence-specific artificial ribonucleases reported to date fall in two categories: (i) oligonucleotide conjugates of metal ion chelates that catalyze the hydrolysis of a phosphodiester bond(s) of the RNA target [28-30] or cleave RNA by a radical-induced degradation of a ribose moiety [31,32], and (ii) metal-independent oligonucleotide conjugates of peptide-like oligomers [33-37]. Although some metal-dependent artificial ribonucleases display high ribonuclease activity in vitro [38-41] they suffer from a number of disadvantages, e.g. uncontrolled diffusion of radicals, which creates the risk of degradation of nontarget biopolymers.
The reduced specificity of metaldependent artificial ribonucleases, arising from difficulties in controlling their hydrolytic activity, represents the main drawback for this approach and encourages development of metal-independent RNA-cleaving ribonuclease biomimetics, capable of cleaving RNA efficiently without additional exogenous cofactors (i.e., metal ions, oxygen, redox reagents). For the last few years a number of such metalindependent artificial ribonucleases has been designed and synthesized [33-37]. However, their hydrolytic activity remains inferior to that of metal-ion dependent artificial nucleases. In addition, most of engineered sequence-specific artificial ribonucleases suffer from limited or zero catalytic turnover. Therefore, one of the challenges in designing chemical ribonucleases is to find an appropriate balance between high efficacy and specificity in terms of RNA cleavage.
Peptidyl Oligonucleotide Chemical Ribonucleases
Recently, a new type of chemical ribonucleases [42-48], showing very unusual catalytic and structural properties, was discovered. These novel oligonucleotidemediated chemical nucleases were constructed by chemical conjugation of short, catalytically inactive oligopeptides containing a regular arrangement of alternating basic (Arg and/or Lys) and hydrophobic amino acid residues (Leu and/or Ala) with an oligonucleotide component (4 – 9 nucleotide residues), which was poorly or non-complementary to RNA target regions (Figure 3).
Figure 3: Schematic representation of peptidyl-oligonucleotide chemical ribonucleases.
It has been reported that these oligonucleotide conjugates bearing peptide groups at the 5’-terminal phosphate, cleave RNA phosphodiester bonds very effectively [42-48]. According to some reports [42, 47], the peptidyloligonucleotide conjugates containing the oligonucleotide recognition element, which is complementary to the target region of the RNA sequence, may catalyze a site-directed cleavage of the regions adjacent to the hybridization site. In addition, the substantial cleavage of other RNA sites was detected by these conjugates, which were located distantly from the major RNA binding region. The other reports [43, 44-46, 48] provided evidence that efficient RNA cleavage can also be achieved by peptidyloligonucleotide conjugates containing ‘random’ oligonucleotide elements, which are poorly or non-complementary to the RNA target. The cleaving potential of the studied conjugates was demonstrated using an in vitro transcript of human tRNALys<sub>3</sub>, tRNAPhe, a 96mer fragment of HIV-1 RNA (123 – 218 nt) comprising the primer binding site and short synthetic RNA sequences [42, 43, 45-48]. At one extreme a direct site-specific cleavage of the RNA target was observed with the fully complementary oligonucleotide sequence without significant catalytic amplification [42, 47]. At the other extreme [43, 44-46, 48], a more efficient RNA cleavage (up to 80% in 24 hr) coupled with a high catalytic turnover was achieved using peptidyl-oligonucleotide conjugates containing oligonucleotide sequences, which were poorly or non-complementary to the RNA target.
The most remarkable feature of these novel biocatalysts was that the covalently attached oligonucleotide units induced catalytic activity of a previously inactive peptide and modulated its cleavage specificity towards RNA. The GpX cleavage specificity was reported for the first time for artificial chemical nucleases. Furthermore, the conjugates exhibited a reaction catalytic turnover of up to 175 in 24 hours [46]. Moreover, for some conjugates rate enhancement of the cleavage of phosphodiester bonds reached up to 107–108 fold over spontaneous RNA hydrolysis [48]. It was demonstrated that both the efficiency of the cleavage and the RNA cleavage patterns depended on the sequences and secondary structure of the peptide and oligonucleotide units, providing artificial enzymes with aptamerlike properties [48].
The preliminary structural studies of these chemical nucleases performed by Circular Dichroism (CD) [45] and our 2D NMR (Bichenkova et al. unpublished data) revealed that both oligonucleotide and peptide components cross-modulate each others’ conformations leading to a formation of a new entity with unique structural and functional properties. The oligonucleotide component seems to induce an `active` conformation of the peptide, and hence, significantly enhance its catalytic performance.
However, the main challenge of combining sequence specificity with high catalytic turnover remains unmet for this class of artificial ribonucleases and could potentially be resolved by identifying structural rules of the molecular mechanisms governing biochemical activity of these highly efficient biocatalytic systems.
Conclusive Remarks and Future Directions
In spite of the advances made toward the development of sequence-specific artificial ribonucleases, the problem of insufficient efficiency of the existing metalindependent chemical ribonucleases, their poor selectivity and low catalytic turnover remains unmet. Therefore, further progress in the search for more efficient and selective ribonucleases will depend on the biophysical and structural studies as well as on the development of novel synthetic strategies to provide new structural variants of cleaving constructs in order to ensure effective and specific RNA cleavage.
Although some impressive advances have been recently achieved in this area due to the recent discovery of novel peptidyloligonucleotide chemical ribonucleases [42-48], neither structural aspects of their ‘active’ conformation(s) nor the modes of their interactions with the RNA sequences have yet been studied. The great challenge is therefore to provide understanding at the molecular level of how these functionally significant entities (i.e., peptide and oligonucleotide) interact with each other within the conjugate and cross-modulate their activities. The lack of knowledge of the structural aspects and the mechanisms involved in the cross-modulated conformational rearrangement within the peptidyl-oligonucleotide chemical ribonucleases, which seem to underpin their catalytic performance, hinders the development of a new generation of peptidyl-oligonucleotide conjugates. Also, very little is known about the modes of interactions between peptidyloligonucleotide chemical ribonucleases and target RNA sequences, which create the additional barrier for improved design of new structural variants of peptidyloligonucleotide chemical ribonucleases with an appropriate balance between sequence-specificity and high catalytic turnover.
Finally, multi-specific targeting of RNA using selected overlapping regions of mRNA transcripts encoding disease-relevant proteins would allow more efficient translational arrest and thus would give more chance of success. This approach, which is expected to offer a synergistic effect, has already been successfully demonstrated in human prostate PC3 cells [49].
References
- Crooke S.T. Annu Rev. Med. (2004), 55, 61-95.
- Henke E., Perk J., Vider J., Candia P., Chin Y., Solit D.B, Ponomarev V., Cartegni L., Manova K., Rosen N. and Benezra R. Nature Biotechnology (2008), 26 (1) 91-100.
- Sun X., Kanwar J. R., Leung E., Lehnert K., Wang D. and Krissansen G. W., Gene Therapy (2001), 8, 638-645.
- Gleave M.E. and Monia B.P. Nature Reviews /Cancer (2005), 5, 468 – 479.
- Elez R., Piiper A., Giannini C.D., Brendel M., Zeuzem S. Biochem. Biophys. Res. Com. (2000) 269: 352-356.
- Dachs G.U., Dougherty G.J., Stratford I.J. and Chaplin D.J. Oncology Research (1997), 9, 313-325.
- Mateo-Lozano S., Gokhale P.C., Soldatenkov V. A., Dritschilo A., Tirado O.M., Notario V. Clin.Cancer Res. (2006), 12(22), 6781-6790.
- Greenberger L.M., Horak I. D., Filpula D., Sapra P., Westergaard M., Frydenlund H. F, Albæk C., Schrøder H., Ørum H. Mol. Cancer Therapy (2008), 7(11), 3598-3608.
- Trawick B.N., Daniher A.T., and Bashkin J.K. Chem. Rev. (1998), 98, 939-960.
- M. Lipsitch, Robins, J.M., Mills, C.E., Bergstrom, C.T. Science (2006), 312, 845.
- Krug R.M. Science (2006), 311, 1562-1563.
- Clercq E. De. Nat. Rev. Drug. Discov. (2007), 6, 1001-1018.
- Clercq E. De. Nat.Rev.Microbiol. (2004), 2, 704-720.
- Clercq E. De. J.Clin.Virol. (2004), 30, 115-133.
- Sullenger B.A. and Gilboa E. Nature (2002), 418, 252-258.
- Stenvang J. and Kauppinen S. Expert Opin. Biol. Th. (2008), 8, 59-81.
- Chow C.S., and Bogdan F.M. Chem. Rev. (1997), 97, 1489-1513.
- Zhang, Y.C., Taylor M.M, Samson W.K., Phillips M.I. Methods. Mol. Med. (2005), 106, 11-34.
- Crooke, S.T. Med. Res. Rev. (1996), 16 (4), 319-344.
- Crooke, S.T. BBA-Gene struct. Expr. (1999), 1489 (1), 31-43.
- Crooke, S.T. Oncogene (2000), 19 (56), 6651-6659.
- Johannes L. and Decaudin D. Gene Therapy (2005), 1–9.
- Arnold U. and Ulbrich-Hofmann R. Biotechnol. Lett. (2006), 28, 1615–1622.
- Sioud M. Trends in Pharmacol. Sci. (2004), (1), 22-28.
- Grimm D. and Kay M.A. J. Clin. Invest. (2007), 117(12), 3633–3641.
- Masiero M., Nardo G., Indraccolo S., Favaro. E. Mol. Asp. Med. (2007), 28, 143-166.
- Sibley C.R, Seow Y. and Wood M.J.A. Mol. Ther. (2010), 18 (3), 466-467.
- Hall, J. Husken, D., Haner R. Nucleosides Nucleotides. (1997), 16, 1357-1368.
- Huang L., Chappell L.L., Iranzo O., Baker B.F., Morrow J.R. J. Biol. Inorg. Chem. (2000), 5, 85-92.
- Sakamoto A., Tamura T., Furukawa T., Komatsu Y., Ohtsuka E., Kitamura M., Inoue H. Nucl. Acids Res. (2003), 31, 1416-1425.
- Sigman D.S.; Mazumder A., Perrin D.M. Chem. Rev. (1993), 93, 2295-2316.
- Duarte V., Sixou S., Favre G., Pratviel G., Meunier, B. J. Chem. Soc., Dalton Trans. (1997), 21, 4113-4118.
- Komiyama M. and Inokava T. J. Biochem. (1994), 116, 719–720.
- Yurchenko L., Silnikov V., Godovikova T., Shishkin G., Toulme J.-J. Vlassov V. Nucleosides Nucleotides, (1997), 16, 1741–1745.
- Ushijima K. and Takaku H. Biochim. Biophys. Acta. (1998), 1379, 217–223.
- Verbeure B.; Lacey C.J., Froeyen M., Rozenski J., Herdewijn P. Bioconjugate Chem. (2002), 13, 333-350.
- Beloglazova N.G., Fabani M.M., Zenkova M. A., Bichenkova E.V., Polushin N.N., Sil’nikov V.N., Douglas K.T., and Vlassov V.V. Nucl. Acids Res. (2004) 32 (13), 3887–3897.
- Morrow J.R., Buttrey L.A., Shelton V.M., Berback K.A. J. Am. Chem. Soc. (1992), 114, 1903-1905.
- Magda D., Miller R.A., Sessler J.L., Iverson B.L. J. Am. Chem. Soc. (1994), 116, 7439-7440.
- Häner R., Hall J., Pfützer A., Hüsken D. Pure Appl. Chem. (1998), 70, 111-116.
- Canaple L., Husken D., Haner R. Bioconjugate Chem. (2002), 13, 945-951.
- Mironova N.L., Pyshnyi D.V., Ivanova E.M., Zarytova V. F., Zenkova M. A., Gross H. J., Vlassov V. V. Russ Chem Bull. (2002), 51, 1177-1186.
- Mironova N.L., Pyshnyi D.V., Ivanova E.M. Nucleic Acids and Molecular Biology. (2004), 13, 151-172.
- Mironova N.L., Pyshnyi D.V., Ivanova E.M., Zenkova M.A., Gross G.J., Vlasov V.V. Dokl. Biochem. Biophys. (2002), 385,196-200.
- Mironova N.L., Pyshnyi D.V., Stadler D.V., Prokudin I.V., Boutorine Y.I., Ivanova E.M., Zenkova M.A., Gross H.J., Vlassov V.V. J Biomol Struct Dyn. (2006), 23, 591-602.
- Mironova N.L., Pyshnyi D.V., Ivanova E.M., Zenkova M.A., Gross H.J., Vlassov V.V. Nucleic Acids Res.(2004), 32, 1928-1936.
- Pyshnyi D., Repkova M., Lokhov S., Ivanova E., Venyaminova A., Zarytova V. Nucleosides & Nucleotides. (1997), 16,1571-1574.
- Mironova N.L., Pyshnyi D.V., Shtadler D.V., Fedorova A.A., Vlassov V.V., Zenkova M.A. Nucl Acids Res. (2007), 35, 2356-2367.
- Gautschi O., Tschopp S., Olie RA., Leech SH., Simões-Wüst AP., Ziegler A., Baumann B., Odermatt B., Hall J., Stahel RA., Zangemeister-Wittke U. Journal of the National Cancer Institute, (2001), 93, 463-471.
This article was printed in the August/September 2011 issue of International Drug Discovery,6,, 4,. Copyright rests with the publisher.