Abstract
Kinase inhibitors are important medicines for the treatment of several types of cancers that harbor dominant genetic lesions in specific kinases. Indeed, tumors with activating mutations, amplifications or translocations of kinases, often respond dramatically to treatment with kinase inhibitors. However, two recurrent themes are emerging based on clinical experience with kinase inhibitors: one is that some tumors with well-characterized genetic lesions in kinases can be intrinsically resistant to kinase inhibitors, that is, they never respond to treatment and two, most tumors that do respond acquire resistance during the course of treatment. The identification of several intrinsic and acquired resistance mechanisms has opened the door to the development of next-generation kinase inhibitors or to alternative strategies to treat resistant tumors. I will discuss examples of resistance mechanisms and discuss future directions for treatment strategies aimed at prolonging the life span of cancer patients treated with kinase inhibitors.
Kinases, many of which are oncogenes, are signaling enzymes that play critical roles in cell growth and survival. Because of that, they have been the object of intense research to identify their specific role in cancer and to discover compounds that inhibit their activity. The past two decades have generated many successes in both endeavors. These include the appreciation that kinases can play a dominant role in certain cancers as a consequence of genetic lesions such as point mutations, amplification or chromosomal translocation that aberrantly activate the kinase. Such dominance by a given kinase in cancer cells has been dubbed “oncogenic addiction”[1]. This term implies that cancer cells are dependent on the function of the kinase for their survival, and by the same token, the “addicted” cells are quickly eliminated when the kinase is inhibited. The second, more important success was achieved in the clinic. Indeed, inhibitors of dominant kinases such as ABL in BCR-ABL positive chronic myelogenous leukemia (CML), EGFR in non-small cell lung cancer (NSCLC) with EGFR activating mutations, c-Kit in gastro-intestinal stromal tumors (GIST) with c-Kit activating mutations, BRAF in BRAFV600E positive melanoma, and ALK in NSCLC with ALK translocations, have been successful drugs to treat these cancers.
c-Met is one of few kinases that is thought to harbor dominant genetic lesions at a reasonable frequency in human cancers, but for which no treatment has been approved. Several c-Met kinase inhibitors are being tested in clinical trials in cancer patients, but there is no clear proof that tumors addicted to c-Met exist. The preclinical data suggesting that c-Met is a dominant oncogene in certain tumor types is, however, very clear. c-Met can play a dominant role in cancer as a consequence of a genetic lesion in the MET gene. Indeed, cancer cell lines with MET amplification can be highly sensitive to a c-Met kinase inhibitor. MET gene amplification has been described in gastric and in lung cancer cell lines. The frequency of MET amplification in gastric and lung cancer patients is expected to be low (5-20%), but not unlike the frequency of EGFR mutations or ALK translocations in lung cancer. MET activating mutations have also been described in a few tumor types, but most frequently in hereditary papillary renal cancer (HPRC).
The Emergence of Resistance to Kinase Inhibitors
While many patients with the appropriate tumor profile respond dramatically to kinase inhibitors, the clinical response is too often short-lived, lasting several months instead of the initial hope of many years. Resistance to kinase inhibitors is now a well-recognized phenomenon that has been observed with all clinically effective kinase inhibitors. Though not a unique phenomenon with kinase inhibitors, drug resistance has been exceptionally well-documented for this class of drugs, perhaps due to their relatively straight forward mechanism of action compared to other anti-cancer drugs, and perhaps also due to the dramatic initial anti-tumor effects that prompted investigations to elucidate the reasons for tumor recurrence. Tumor recurrence during treatment with a kinase inhibitor was first observed with the first kinase inhibitor approved for cancer treatment, imatinib. The fact that CML is a disease amenable to the acquisition of multiple tumor samples enabled rapid bedside-to-bench experimentation to identify mechanisms of drug resistance. Several important discoveries were made that turned out to be relevant for other kinases and their inhibitors. For example, novel mutations in the kinase domain were discovered that prevented drug binding or decreased the affinity of the drug for its target. One particular mutation at the “gatekeeper” residue of the kinase domain appears to be a common mechanism of resistance. Indeed, gatekeeper residue mutations have been found in ABL, EGFR, ALK and c-Kit after tumors become resistant to their respective inhibitors. While the duration of response, especially in solid tumors, has not been as long as was hoped for, the ability to obtain tumor samples after disease recurrence has made further research into the causes of drug resistance possible.
Intrinsic vs. Acquired Resistance
It is important to distinguish between two types of resistance to kinase inhibitors that is encountered in pre-clinical models and in clinical practice: intrinsic resistance and acquired resistance. Intrinsic resistance refers to tumors that do not respond at all to kinase inhibitors and is in contrast to acquired resistance which refers to tumors that initially responded well to treatment, but for which growth resumed at some point in time during treatment. Intrinsic resistance, itself, can be considered in two ways when discussing kinase inhibitors. For example, most tumors are intrinsically resistant to kinase inhibitors because they do not contain dominant genetic lesions in kinases. Secondly, tumors that harbor a genetic lesion that is thought to be usually dominant can also fail to respond to kinase inhibitors. Today, it is common practice in many large cancer centers to prospectively identify which cancer patients harbor dominant kinase mutations in order to treat them appropriately. This novel approach avoids treating most patients that would be intrinsically resistant to therapy because they lack the appropriate genetic characteristics.
Mechanisms of Intrinsic Resistance
The greatest opportunity to identify mechanism of intrinsic resistance is in those patients that harbor the correct dominant genetic lesion, but still fail to respond to treatment. The tumor biopsies obtained for initial diagnosis could also be used to test for concurrent mutations in other genes that may account for the intrinsic resistance. Pre-clinical models could also be used to assess potential mechanisms of intrinsic resistance. For example, some lung cancer cell lines with MET amplification are intrinsically resistant to c-Met inhibitors. In one case, a KRAS mutation is also present which may explain the intrinsic resistance. Such pre-clinical examples offer a model system to identify possible ways to overcome resistance.
One important question is whether some kinases that lack mutations, amplifications or translocations still may play a role under some circumstances. In such a scenario, one could imagine that combinations of kinase inhibitors could be useful. For example, c-Met and EGFR can be coactivated in lung cancer cell lines, even if both are wild-type. This observation offers the possibility that inhibition of both c-Met and EGFR may be effective in lung cancer, but this will need to be tested.
A better understanding of mechanisms of intrinsic resistance should open the door to more effective therapies. However, it is anticipated that these mechanisms will be varied and that much more research will be required before clinical progress can be made.
Mechanisms of Acquired Resistance
As stated above, essentially all tumors initially sensitive to kinase inhibitors become resistant over time, thus making clear that kinase inhibitors exert selective pressures on tumors to find escape routes. The escape mechanisms can be different based on the tumor type, but also based on the characteristics of the inhibitor.
The most common mechanism leading to acquired resistance is the acquisition of a secondary mutation in the kinase that prevents drug binding or lowers the affinity of the drug. Multiple such mutations have been described in different cancers treated with different kinase inhibitors. The attempt to overcome this mechanism of resistance has met with some success so far. Patients with resistant tumors have been treated with second-generation kinase inhibitors that are active on some secondary mutations. This exciting finding illustrates the important point that different inhibitors against the same kinase can become useful follow-on treatments
c-Met offers another interesting example related to acquired resistance. c-Met small molecule kinase inhibitors can be divided into two broad classes, Class I and Class II, based on their binding mode to the kinase domain of c-Met. The two classes also differ in their selectivity for other kinases and in their ability to inhibit different mutants of c-Met. Class I inhibitors are very selective for c-Met and do not bind to other kinases while class II molecules are multikinase inhibitors. On the other hand, class II molecules can inhibit several mutants of c-Met, but class I inhibitors show much weaker activity for some c-Met mutants compared to the wild-type c-Met kinase. Because of the differences in the selectivity and in the ability to inhibit different c-Met mutations, it is not unreasonable to expect that class I and class II c-Met inhibitors would 1) exert different selection pressure on tumors and therefore lead to different resistance mechanisms and 2) that each class may find utility in the clinic by having a different pattern of activity on wild-type and mutant c-Met.
Another resistance mechanism that is emerging occurs through the activation of alternative pathways to compensate for the loss of signaling from the dominant kinase. For example, gene ampliation of the kinase MET was observed in pre-clinical studies and in a small number of NSCLC patients as an acquired resistance mechanism to erlotinib treatment. Activation of other receptor tyrosine kinases such as IGF-1R, AXL and PDGFR has also been documented [2]. These data demonstrate that tumors that are addicted to signaling pathways can adapt to pathway inhibition by switching their signaling requirements to other pathways. Such flexibility of tumors to adapt suggests that multiple pathways should be blocked as an initial course of therapy rather than relying on a single drug.
Challenges and Opportunities
The main challenge for the development of second-generation kinase inhibitors is the fact that the patient population served by many of these drugs is relatively small (for example, EGFR mutant NSCLC). On the other hand, since screening for the presence of genetic lesions is becoming more common at various stages of the disease, it will become easier to identify patients that should receive treatment aimed at resistant tumors. The above challenge vs. opportunity should be kept in mind with the discussion below.
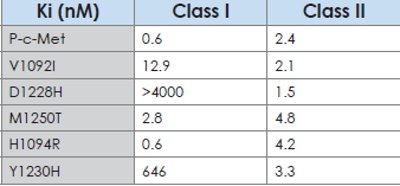
Table 1-Activity of class I and class II c-Met inhibitors on wildtype and mutant c-Met in an in vitro kinase assay. Class II inhibitors have similar activity on wild-type and on several mutants whereas some mutants are less sensitive or potentially resistant to class I compounds. This example illustrates that different classes of c-Met inhibitors may be useful under different circumstances.
Anticipate seconday mutations: since secondary mutations have been observed with multiple kinase inhibitors against different targets, it suggests that this particular mechanism of resistance is likely to occur with other kinase inhibitors. It therefore makes sense to develop novel kinase inhibitors that will inhibit the wild-type and several mutants in the same molecule if possible. Which mutations are likely to occur in patients cannot be completely predicted, but pre-clinical studies in which cell lines were made resistant to kinase inhibitors have been generally useful.
Whereas some kinase mutations can be very resistant to a given kinase inhibitor, some other mutations can be somewhat resistant. in this case, it is conceivable that higher doses of the inhibitor could overcome resistance. This has been demonstrated with imatinib in CML [3]. While this appears as a simple solution, this would be achievable only with molecules that are extremely safe as toxicities will limit the amount of drug a patient can tolerate. in anticipation of this possibility, future kinase inhibitors should be carefully chosen based on better safety profi le. This is usually associated with kinase inhibitors that show a greater degree of selectivity against all other kinases. This concept can be illustrated with the two classes of c-Met inhibitors (Figure). it is anticipated that class i inhibitors may be better tolerated than class ii molecules because they are highly selective. if this turns out to be the case, it is possible that class i inhibitors may be active in vivo against wild-type c-Met as well as against some less sensitive mutants of c-Met. The challenge in the above scenario is two-fold: one, it can be very diffi cult and perhaps not feasible in some instances to develop very selective inhibitors for some kinases and two, even very selective inhibitors can come with side effects if the kinase of interest plays an important role in normal physiology.
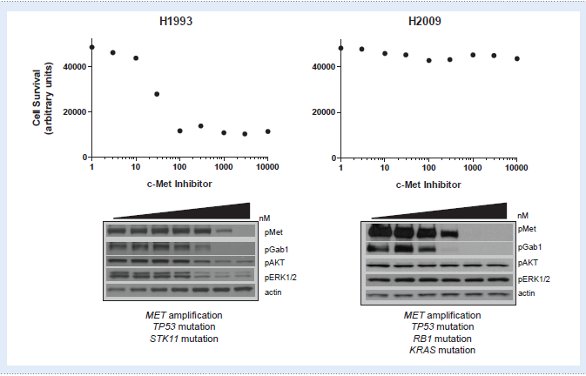
Figure 1 - H2009 lung cancer cells are intrinsically resistant to a c-Met inhibitor despite the presence of MET gene amplifi cation. Top: H1993 lung cancer cell lines are sensitive to a c-Met inhibitor in a 72-hour survival assay while H2009 show no effect. Bottom: Signaling pathways are sensitive to a c-Met inhibitor in H1993 cells. In H2009, c-Met and its direct substrate, GAB-1, are sensitive to a c-Met inhibitor, but other pathways downstream are resistant. Other mutations, such as a KRAS mutation, may account for the intrinsic resistance in H2009 cells.
Signaling fl exibility as a challenge and an opportunity: many examples exist, at least pre-clinically, where cancer cells can reinitiate or re-direct signaling important for their growth and/or survival as a result of treatment with a kinase inhibitor. Because signaling is very fl exible in that there are many opportunities to connect to important pathways, it will be challenging to 1) identify which pathway is most often used as a by-pass mechanism and 2) completely block access to these pathways with one inhibitor. For these reasons, combination therapy with multiple kinase inhibitors seems to be a possible solution. This will require “best-in-class” molecules that are potent, selective and safe if kinase inhibitors are to be combined successfully.
The Future of Kinase Inhibitors in Cancer Treatment
There are multiple inhibitors for different kinases that have been approved for the treatment of various cancers and many more are being tested in the clinic. some could argue that kinase inhibitors have not been as successful as originally anticipated, that is the percentage of patients that can benefi t from kinase inhibitors is relatively small. However, what kinase inhibitors have done for cancer patients and for basic and clinical cancer research in the past decade is extraordinary: they helped defi ne new drivers of cancers, they enabled biomarker implementation, they pushed the barrier of tumor sample collection and use, they have helped defi ne subgroups of cancer patients within the same disease. There have been many challenges along the way and many more will need to be overcome. Drug resistance is probably the biggest challenge right now. Clinicians’ increased ability to obtain pre-treatment and postrecurrent tumor biopsies has enabled the study of clinically relevant drug resistance mechanisms and will continue to be an essential iterative process of bedside-tobench research critical for the discovery of novel approaches to combat drug resistance.
There will be resistance mechanisms that do not involve kinases and this is the next challenge that we will need to tackle. However, the instances where kinases are involved are technically within our reach and there is no reason why we should not solve that problem – it is within our reach.
References
- Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63-4.
- Xu Y, Liu H, Chen J, Zhou Q. Acquired resistance of lung adenocarcinoma to EGFR-tyrosine kinase inhibitors gefi tinib and erlotinib. Cancer Biol Ther. 2010; 9:572-82.
- Jabbour E, Kantarjian HM, Jones D, Shan J, O’Brien S, Reddy N, et al. Imatinib mesylate dose escalation is associated with durable responses in patients with chronic myeloid leukemia after cytogenetic failure on standard-dose imatinib therapy. Blood. 2009;113:2154-60.
Dr. Dussault is currently Director of Oncology Research at Amgen, Inc. Her experience spans from target identifi cation to clinical candidate identifi cation and fi rst-in-human studies. Her team at Amgen is responsible for the discovery of different types of c-Met small molecule kinase inhibitors and for the identifi cation of their respective potential for the treatment of different cancers.
This article was printed in the 11/14/2011 issue of International Drug Discovery,6,, 5,. Copyright rests with the publisher.