Abstract
The Ten-eleven translocation (TET) protein family is a well-conserved group of proteins consisting of TET1, TET2, and TET3 members that have various roles in gene expression by converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) in the DNA. The impact of these proteins reaches several aspects of human life—including cell growth regulation, embryonic stem cell maintenance, and cell differentiation—as well as a number of mutations leading to a multitude of diseases, such as those induced by chromosomal translocations and those that lead to cancer. Though the TET family of proteins has only recently begun to reveal its far-reaching implications, it has already shown itself as a key player demanding far greater attention from the biomedical research community. This article will discuss TET inhibitors as possible novel anti-cancer drugs. We will also discuss how inhibitors of metabolic enzymes, such as fumerate hydratase (FH), isocitrate dehydrogenase (IDH), and succinate dehydrogenase (SDH), might also be new avenues for anti-cancer drug research. These enzymes are in the citric acid cycle, are frequently mutated in cancer, and lead to the production of alpha ketoglutarate (α-KG) which is used as a cofactor for TET and is required for its activity.

Introduction
Environmental factors can alter the way our genes are expressed, making even identical twins—with their identical DNA sequences—different. Epigenetics refers to these heritable patterns of gene expression that arise with the absence of DNA sequence alteration and is largely dictated by regulated chromatin structure, including covalent modifications of histones and cytosine methylation [1]. The information contained in eukaryotic DNA is stored and expressed in tightly controlled ways, primarily through post-replicative methylation of DNA and post-translational modifications of histones [2]. These modifications are collectively known as epigenetic marks, which regulate chromatin organization and gene expression patterns without altering the primary nucleotide sequence of DNA [2].
The best characterized epigenetic mark is a methyl group at the 5-position of cytosine bases known as 5-methylcytosine (5mC) (Fig. 1). 5mC has been studied extensively, and its role as an epigenetic modification involved in gene regulation X-chromosome inactivation, genomic imprinting, long-term silencing of transposons, and cancer development is well described [2]. Cytosine exists as a free nucleotide that is incorporated into DNA during replication. The pattern of 5mC in the genome is accurately preserved by mitotic inheritance through the action of DNA methyltransferases (DNMTs), specifically the maintenance DNA methyltrasferase DNMT1, which catalyze the covalent addition of methyl groups to cytosine in newly synthesized DNA [3]. 5mC is formed by post-replicative addition of a methyl group to cytosine through the action of DNA methyltransferases (DNMTs), which use S-adenosyl methionine (SAM) as the methyl donor [3].
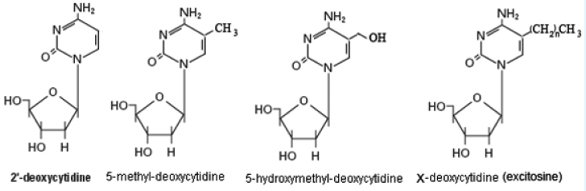
Figure 1: The many faces of cytosine. The “excytosine” modification refers to as-to-yet undiscovered modifications to cytosine that are resistant to bisulfite treatment.
5-hydroxymethylcytosine (5hmC) was discovered in mammalian DNA in 1972 (Fig. 1) [4], but it was thought to be an oxidative-damage product of DNA of little importance and was therefore largely ignored. 5hmC was re-discovered in 2009 by the laboratories of Nat Heintz [5] at Rockefeller University and Anjana Rao at Harvard Medical School [6], and became of interest when it was shown that the TET family of dioxygenases utilize molecular oxygen to transfer a hydroxyl group to 5mC in the catalytic conversion of 5mC to 5hmC, and that one of these enzymes, TET2, is frequently mutated in myeloid neoplasms [7, 8]. The formation of 5hmC can lead to demethylation of DNA, which may contribute to the dynamics of DNA methylation [9]. The recent evidence linking TET to aberrant DNA methylation has revealed that oxidation of 5mC occurs in human cells, catalyzed by TET proteins, and the resulting 5hmC may have opposite or alternative functions as 5mC. We speculate that there are as yet undiscovered modifications of cytosine, and possibly other bases, that have yet-to-be-determined epigenetic effects during development (Fig. 1, we refer to 5xC as “excytosine”).
Dense methylation found around gene promoters in so-called CpG islands is associated with gene silencing; thus, the distribution of methyl groups in the genome defines regions of varying transcriptional potential [10]. The DNA methylation during embryonic development results in tissue- specific patterns, which may explain the connection to important aspects of cellular differentiation [11].
Cytosine methylation has important roles in key biological processes, including several gene-regulatory mechanisms. Mechanisms responsible for X-chromosome inactivation in females, allele-specific silencing of imprinted genes, and transcriptional repression of transposons is just a few that have been implicated [10]. Cytosine methylation also causes a number of human syndromes when there is disruption of methylation status in imprinted genes [12, 13] or mutations in genes encoding components of the DNA-methylation machinery [14, 15]. Alterations in DNA methylation patterns have been implicated in autoimmune diseases [16], neurological and psychiatric conditions [17-19], aberrant stem cell development, and cancer [20-22].
Amongst the negative impacts relevant to aberrant DNA methylation, cancer is one of the most studied afflictions where causes are not well understood. The genomes of tumor cells are generally characterized by a global loss of methylation (hypomethylation), accompanied by focal increases in methylation (hypermethylation) [20-22]. Global hypomethylation may lead to genomic instability (a hallmark of cancer) [23], and hypermethylation of gene promoters may lead to transcriptional silencing of tumor suppressor genes [20-22]. Other methylation events may affect tissue-specific differentiation, which, according to a prevailing model , is the main mechanism by which epigenetic changes cause cancer [24].
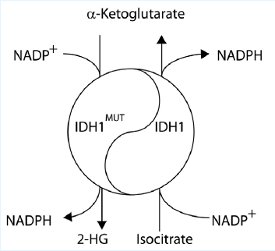
Figure 2: The Yin-Yang of Isocitrate Dehydrogenase. The wild-type IDH1 produces a-ketoglutarate which is used by TET. The IDH1MUT produces 2-HG which inhibits TET and thereby decreases global 5hmC levels.
Recent genomic sequencing efforts in acute myeloid leukemia (AML) and in other malignancies have identified new classes of oncogenic disease alleles. One recently identified class of genes mutated in cancer are those coding for enzymes involved in citrate metabolism. The most prevalent of such mutations identified to date affect the genes for cytosolic isocitrate dehydrogenase 1 (IDH1) and its mitochondrial homolog IDH2 [25, 26]. Other metabolic mutants associated with cancer are succinate dehydrogenase (SDHD) [27, 28] and fumarate hydratase (FH) [29]. These findings have made metabolism, and drugs that affect metabolism, as one of the most important areas of cancer research and drug discovery [30].
IDH1 and IDH2 are NADP+-dependent enzymes that normally catalyze the interconversion of isocitrate and alpha-ketoglutarate (α-KG; also known as 2-oxoglutarate). A dehydrogenase (also called DHO in the literature) is an enzyme that oxidizes a substrate by transferring one or more hydrides (H) to an acceptor. An oxygenase is any enzyme that oxidizes a substrate by transferring the oxygen from molecular oxygen O2 (as in air) to it. Dioxygenase is an oxygenase that transfers both of the oxygen atoms in O2 to the substrate. Two distinct alterations are caused by the tumor-derived mutations in IDH1 or IDH2: loss of its normal catalytic activity in the production of a-ketoglutarate (α-KG) and gain of the catalytic activity to produce 2-hydroxygulatrate (2-HG) (Fig. 2) [25, 26].
IDH1R132MUT is a gain of function mutation in isocitrate dehydrogenase that produces the “cancer metabolite” 2-hydroxyglutamate (2-HG), which inactivates TET1. 2-HG is a competitive inhibitor of multiple α-KG-dependent dioxygenases, including histone demethylases and the TET family of 5-methylcytosine (5mC) hydroxylases. The NADP+-dependent isocitrate dehydrogenase genes IDH1 and IDH2 are mutated in >75% of low-grade gliomas and secondary glioblastoma multiforme (GBM) and in ~20% of AML leukemia [25, 26]. When its normal catalytic activity is lost, mutant IDH1 and IDH2 also gained the function of catalyzing the reduction of a-KG to produce 2-hydroxyglutarate (2-HG), resulting in an accumulation of D-2-HG (also known as R-2-HG) in IDH1 or IDH2 mutated gliomas and AML. In IDH1 mutated glioma, D-2-HG accumulated to extraordinarily high levels of 5–35 mmol/g of GBM [31], which could be equivalent to 5–35 mM assuming the tissue density of 1 g/ml.
IDH1 and IDH2 mutations were subsequently observed in myeloid malignancies including de novo and secondary AML (15%–30%) and preleukemic clonal malignancies including myelodysplasia and myeloproliferative neoplasms (5% of chronic phase and 20% of transformed cases) [32, 33]. The precise genetic context in which IDH1/2 mutations occur is not known, nor is the mechanism through which they contribute to the malignant phenotype. The most common IDH1/2 mutations in AML and brain tumors, affecting R132 of IDH1 or R140 and R172 of IDH2, have the common feature of acquiring a neomorphic enzymatic activity catalyzing the NADPH-dependent reduction of αKG to R(−)-2-hydroxyglutarate (2HG).
The genetic connection has given a major boost to the field of cancer metabolism. Normal cells primarily generate energy aerobically in mitochondria, whereas most tumor cells rely more heavily on glycolysis (the anaerobic conversion of glucose to lactate) to generate energy. Tumors also display other unique metabolic features. But the discovery of a mutated metabolic enzyme was strong evidence that metabolic abnormalities play an important role in oncogenesis, (and it set off a frantic effort to understand what the mutation was doing in such common and lethal cancers).
Although the functions of these TET proteins have only begun to be investigated, they are already recognized as major players in molecular medicine. Equally as important, therefore, is the race to now screen for and find TET inhibitors to ameliorate the damaging effects of these proteomic malfunctions. The IDH1 R132MUT mutation has been associated with approximately 30% of glioma tumors and will provide the basis for a novel TET inhibitor screen. Another novel approach for a screening method involves targeting epigenetic enzymes for cancer therapy. All together, the hope is in creating novel drug discovery methodology leading ultimately to better understanding and therapy for patients with TET protein malfunctions.
References
- Bird, A., Perceptions of epigenetics. Nature, 2007. 447(7143): p. 396-8.
- Portela, A. and M. Esteller, Epigenetic modifications and human disease. Nature Biotechnology, 2010. 28(10): p. 1057-68.
- Goll, M.G. and T.H. Bestor, Eukaryotic cytosine methyltransferases. Annu Rev Biochem, 2005. 74: p. 481-514.
- Penn, N.W., et al., The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. The Biochemical journal, 1972. 126(4): p. 781-90.
- Kriaucionis, S. and N. Heintz, The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science, 2009. 324(5929): p. 929-30.
- Tahiliani, M., et al., Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science, 2009. 324(5929): p. 930-5.
- Dahl, C., K. Gronbaek, and P. Guldberg, Advances in DNA methylation: 5-hydroxymethylcytosine revisited. Clin Chim Acta, 2011. 412(11-12): p. 831-6.
- Abdel-Wahab, O., Genetics of the myeloproliferative neoplasms. Current opinion in hematology, 2011. 18(2): p. 117-23.
- Ndlovu, M.N., H. Denis, and F. Fuks, Exposing the DNA methylome iceberg. Trends in biochemical sciences, 2011.
- Law, J.A. and S.E. Jacobsen, Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature reviews. Genetics, 2010. 11(3): p. 204-20.
- Feng, S., S.E. Jacobsen, and W. Reik, Epigenetic reprogramming in plant and animal development. Science, 2010. 330(6004): p. 622-7.
- Uribe-Lewis, S., et al., Molecular mechanisms of genomic imprinting and clinical implications for cancer. Expert reviews in molecular medicine, 2011. 13: p. e2.
- Lim, D.H. and E.R. Maher, Genomic imprinting syndromes and cancer. Advances in Genetics, 2010. 70: p. 145-75.
- Okano, M., et al., DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 1999. 99(3): p. 247-57.
- Malagnac, F., et al., A gene essential for de novo methylation and development in Ascobolus reveals a novel type of eukaryotic DNA methyltransferase structure. Cell, 1997. 91(2): p. 281-90.
- Teitell, M. and B. Richardson, DNA methylation in the immune system. Clinical immunology, 2003. 109(1): p. 2-5.
- Ratan, R.R., Epigenetics and the nervous system: epiphenomenon or missing piece of the neurotherapeutic puzzle? Lancet neurology, 2009. 8(11): p. 975-7.
- Mehler, M.F., Epigenetics and the nervous system. Annals of Neurology, 2008. 64(6): p. 602-17.
- Jiang, Y., et al., Epigenetics in the nervous system. The Journal of neuroscience : the official journal of the Society for Neuroscience, 2008. 28(46): p. 11753-9.
- Costa, F.F., Epigenomics in cancer management. Cancer management and research, 2010. 2: p. 255-65.
- Esteller, M., Cancer epigenomics: DNA methylomes and histone-modification maps. Nature reviews. Genetics, 2007. 8(4): p. 286-98.
- Jones, P.A. and S.B. Baylin, The epigenomics of cancer. Cell, 2007. 128(4): p. 683-92.
- Chen, R.Z., et al., DNA hypomethylation leads to elevated mutation rates. Nature, 1998. 395(6697): p. 89-93.
- Portela, A. and M. Esteller, Epigenetic modifications and human disease. Nat Biotechnol, 2010. 28(10): p. 1057-68.
- Chowdhury, R., et al., The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Reports, 2011. 12(5): p. 463-9.
- Xu, W., et al., Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell, 2011. 19(1): p. 17-30.
- Baysal, B.E., et al., Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science, 2000. 287(5454): p. 848-51.
- Ricketts, C.J., et al., Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Human Mutation, 2010. 31(1): p. 41-51.
- Tomlinson, I.P., et al., Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nature Genetics, 2002. 30(4): p. 406-10.
- Cairns, R.A., I.S. Harris, and T.W. Mak, Regulation of cancer cell metabolism. Nature Reviews. Cancer, 2011. 11(2): p. 85-95.
- Dang, L., et al., Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature, 2010. 465(7300): p. 966.
- Marcucci, G., et al., IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 2010. 28(14): p. 2348-55.
- Ward, P.S., et al., The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell, 2010. 17(3): p. 225-34.
Ms. Nancy Chia is the laboratory manager of the Epigenomics Core Facility at Wayne State University. She obtained a B.S. degree in Biochemistry & Cell Biology at the University of California at San Diego and was a Research Technician at the Broad Institute in Cambridge, MA.
Mr. Luan Wang is a graduate student in Molecular Toxicology in Dr. Ruden’s laboratory and is scheduled to receive his Ph.D. in 2012. He received a B.S. in Biology and M.S. in Biomedicinefrom Beijing Normal University and a M.Sc. from the University University of Tennessee in Nutrition Sciences.
Dr. Douglas Ruden is the Director of Epigenomics and an Associate Professor in the Institute of Environmental Health Sciences, the C. S. Mott Center, and the Department of Obstetrics and Gynecology at Wayne State University in Detroit, MI. He received two B.S. degrees from Caltech and a Ph.D. from Harvard University.
This article was printed in the 12/20/2011 issue of International Drug Discovery,6,, 6,. Copyright rests with the publisher.