Abstract
RNA interference (RNAi) is a powerful tool for gene silencing. Over the past decade our increased understanding of the molecular mechanisms underlying RNAi has provided an avenue towards utilizing it therapeutically. Of the many RNAi precursors, small interfering RNAs (siRNAs) have been the most successfully translated into the clinic. One of the major issues with RNA-based medicines is the cells’ ability to see and respond via pattern-recognition receptors such as Toll-like receptors (TLRs) and retinoic acid inducible gene 1 (RIG-1), inducing innate and adaptive immune responses. We propose that in certain diseases the combination of gene silencing and appropriate immune activation by so-called ‘bifunctional siRNA’ is a rational therapeutic approach and worthy of further investigation. Here we briefly review the progress in developing siRNA-based therapeutics and its interaction with the immune system, including siRNA design, types of immune activation, RNAi use in cancer and viral infections, as well as pre-clinical applications. We believe that development of immunostimulatory siRNAs provides the means for more potent therapies.
Introduction
Fire and Mello first described RNA interference (RNAi) when they injected into C.elegans a mixture of sense and antisense RNA oligonucleotides that resulted in double-stranded RNA (dsRNA) strands, and found it to be more efficient at gene silencing compared to either the sense or the antisense strands alone [1]. Subsequent work from the Tuschl lab showed that a dsRNA as short as 21bp was sufficient to cause gene silencing [2]. Both these findings provided the basis for what is now known as RNAi.
RNAi involves short dsRNA associated with multi-protein complexes that results in complementary mRNA degradation through catalytic cleavage (Figure 1). Central to the process of RNAi is the Ribonuclease-III enzyme Dicer. Dicer cleaves dsRNA into short, 21- and 22- nucleotides fragments with 2nt 3’-OH overhangs, i.e. siRNAs, identified as sequence-specific mediators of RNAi in the cell’s cytoplasm [3, 4]. Dicer then unwinds these short nucleotides and loads one of the RNA strands into the multiprotein RNA-induced silencing complex (RISC)[5]. SIRNA-loaded RISC (SiRISC) then complements target mRNA, causing Ago-2 to slice the mRNA [6]. As the siRNA guide strand is not destroyed by this process and remains associated with RISC this process is catalytic, carrying out multiple rounds of mRNA cleavage [7, 8].
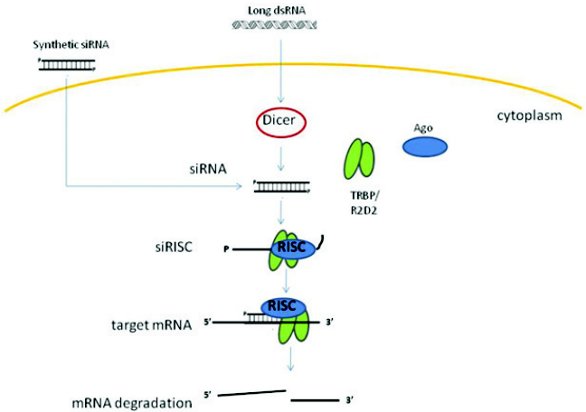
Figure 1: A general schematic overview of the RNAi mechanism. RNA nucleotides are processed by the enzyme Dicer into shorter dsRNA, known as siRNAs. One siRNA strand is loaded into the RISC complex consisting of Ago protein and TRBP/R2D2. SiRISC scans for complementary mRNA and promotes mRNA cleavage. Synthetic siRNAs bypass Dicer processing and can be directly loaded into RISC.
The availability of the complete human genome sequence means that it is trivial to identify targets for gene silencing and design complementary sequences to regulate their expression through RNAi [9-11], thus vastly reducing the development timeline for any potential therapeutic. Furthermore, targeting genes that drive disease, such as Bcl2 in melanoma or the human papillomavirus (HPV) oncogenes E6 and E7 in cervical cancer [12], allows a rational and targeted therapeutic approach via RNAi. However, Dicer is neither the only, nor the first, protein to detect siRNAs within the cell. We now recognize that there is an array of both external and internal RNA-sensing proteins whose role is mainly in anti-microbial responses. It is the ability of siRNA to activate these pathways and the concept that they may offer benefits in the treatment of tumors and viral infections that is the focus of this review.
Types of Immune Activation by siRNAs
It has long been known that long dsRNA results in activation of the interferon response and TLR3 was identified as the dsRNA sensor driving this response [13]. Initially, siRNAs were thought to evade TLR3 activation due to their short nature but subsequently were shown to be potent inducers of innate immune responses [14, 15]. This siRNA-induced immunostimulation occurs via a number of receptors including the RIG-I and/or melanoma differentiation associated gene-5 (MDA-5), PKR, and TLRs-3, -7 and -8 (Figure 2). Each receptor is activated by different motifs present upon the siRNA. For example, a 5’-triphosphate motif activates RIG-I/MDA-5 [16] while the dsRNA-dependent protein kinase (PKR) is activated by long ( (<30bp) dsRNA and siRNAs [14, 15, 17]. Together, these molecules serve as part of the anti-viral response, and therefore, activation of these pathways by siRNAs would be advantageous in targeting virally induced diseases.
SiRNAs are typically recognized by endosomal TLRs-3, -7, and -8 [18, 19]. Ligand binding to TLR results in conformational changes and possibly dimerization, leading to recruitment of crucial adaptor proteins (reviewed in [18]). Engagement of these adaptors activates a series of signal transduction molecules such as myeloid differentiation primary response protein 88 (MyD88), which then activates mitogen-activated protein (MAP) kinases and nuclear translocation of the transcription factor NF-κB, leading to activation of type I IFN genes and subsequently cytokines such as IFNα and TNFα (Figure 2) [20]. Type I IFNs are mediators that bridge the innate and adaptive immune pathways, hence induction of these cytokines is not only able to directly defend against pathogen invasion, but also assist in clearing infected or damaged cells.

Figure 2: SiRNA-induced immunostimulation. Long dsRNA activates PKR to phosphorylate eIF2α, inhibiting protein synthesis. Long dsRNAs as well as 3p-siRNAs activate RIG-I/MDA-5 to trigger innate immunity through transcription of Type I IFNs via IRF3, leading to transcription of IFNα/β through activation of TRIF and its subsequent downstream proteins. Induction of IFNα/β can also occur through actions of MyD88, activated by recognition of siRNAs by TLR7, ssRNA by TLR8, and bacterial CpG DNA/CpG conjugated siRNA by TLR9. MyD88 activation induces IRF7 for transcription of Type I IFN genes. MyD88 adaptor activation also induces NFκB and activates pro-inflammatory cytokines.
We and others have shown in both human and murine systems that siRNAs are recognized by TLR7 via sequence-specific sensing leading to induction of IFNα [21, 22]. TLR7 activation also triggers NF-κB activation, releasing TNFα in cells [23]. SiRNA-induced immunostimulation presented by type I IFNs in cells has also been shown to be important for anti-tumor activity and immunomodulatory effects through activation of natural killer (NK) cells, dendritic cells (DCs), and macrophages, all of which are essential effector cells in the innate immune system [24].
siRNA Immunostimulatory Structure
Traditionally, siRNAs are designed according to criteria known as the Tuschl method; where efficient silencing is obtained with siRNA duplexes composed of 21bp strands, paired in a manner to have 2nt 3’-overhangs, usually consisting of AA or UU nucleotides [25]. Longer siRNA duplexes up to 27bp have also been shown to optimize silencing without activating PKR, which requires at least 30bp to elicit immune activation in a sequence non-specific fashion [26, 27]. However, there has been much debate in the literature about the exact triggers of siRNA-induced immunostimulation. Structural modifications involving addition of CpG-DNA motifs[28], 5’-triphosphate [16] or uridine bulges [21] to siRNAs have been shown to result in increased immunostimulation. Moreover, the amount of uridine in an siRNA duplex influences its immunostimulatory ability. Certain motifs including “UGUGU” and “UXUCU”, have been described that evoke immunostimulation [22, 29]. Judge et al further supported the importance of uridine-based motifs where addition of a string of uridine nucleotides to siRNAs showed an increase of IFNα induction [30]. On the other hand, addition of 2’-O-methyl (2’-OMe) groups to the siRNA backbone abolishes siRNAs immunostimulatory ability. These modifications allow siRNAs to be designed according to a desired function, either by incorporating and increasing siRNA-induced immunostimulation or reducing this immunostimulatory ability.
RNAi and the Immune System
It is clear that the immune system plays a critical role in resisting the formation of cancers. Indeed, immunodeficient patients are more susceptible to cancer formation [31], and mice devoid of adaptive immune responses have higher rates of cancer [32]. A functional immune system is ultimately required for tumor clearance. Indeed, the efficacy of chemotherapeutic agents critically depends on active immune responses [33]. Other than directly targeting negative regulatory genes of the immune system, there are two basic strategies by which one may manipulate the immune system via RNAi. The first is to induce non-specific innate immune responses that are either directly anti-cancer in nature or are required in activating local or systemic effects. The second is to induce specific immune responses against the target antigen.
SiRNA-mediated, Non-specific Immune Responses
SiRNAs are able to activate TLRs to induce inflammatory cytokines such as IFN and TNFα. This would occur both within target tumor cells and cells of the immune system such as resident and circulating macrophages and dendritic cells (DCs), if delivered systemically, leading to activation of innate and adaptive immune responses. Activation of TLRs leads to improved adaptive immune responses by triggering DC maturation, stimulating proliferation of CD4+ and CD8+ T-cells and modulating the suppressive function of Treg-cells [34, 35]. Clinically, such activation is known to improve outcomes in the viral and cancer setting. For example, Imiquimod®, a TLR7 agonist, is approved for use against basal cell carcinomas and HPV genital infections [36]. Furthermore, clinical trials of TLR 7 and 9 agonists have been shown to improve anti-cancer vaccines [37-39]. In animal models the combination of siRNA and immune stimulation has been shown to improve outcomes in the cancer setting. For example, the combination of an siRNA targeting STAT3 with CpG DNA, a TLR9 agonist, gave improved anti-tumor responses [40] while Poeck et al showed that siRNA targeting of Bcl2 in combination with RIG-1 activation gave superior tumor reductions compared to silencing alone [16]. We have shown that combining siRNA silencing of the HPV E6/E7 oncogenes in combination with activation of TLR7 via a single siRNA molecule give significantly better cervical cancer tumor reduction compared to silencing alone [22]. Interestingly, we and others have also noted that merely activating TLRs alone can result in anti-tumor effects [22, 41].
Many of these same cytokines induced by siRNA have been considered for, or are used, clinically as anti-cancer agents. Interferon has been previously used to treat hairy cell leukemia, myeloproliferative disorders, CML, renal cell carcinoma, melanoma and Kaposi’s sarcoma [42-45]. In the anti-viral setting IFN is used to treat Hepatitis B and C [46, 47]. Moreover, these are used at extremely high doses and yet are tolerated by patients. One could argue that the nature and level of the cytokine release induced by siRNAs is more akin to natural activation and thus less likely to cause uncontrolled inflammatory responses. This would be improved further if siRNA delivery can be appropriately directed to specific disease sites such as tumors, using delivery systems.
Much more data exists with regard to utilizing RNAi to combat viral infection. Indeed, RNAi against HIV, Hepatitis B, and RSV infections are currently in different phases of clinical trial [48-50]. However, it should be noted that much effort has been made to remove any immunostimulating activity of these siRNAs, both to be able to account for the direct RNAi effect and to avoid unnecessary inflammation. Yet IFN alone has excellent anti-viral effects, and has been used widely as part of immunotherapy for viral infections, malignant tumors, immunodeficiency and autoimmune disease. Two groups have demonstrated the sequence-specific siRNA recruitment of TLR7 causes anti-viral effects in murine models of influenza detection [51], while activation of TLR7/8 by anti-viral siRNA has been shown to cause reduction of Hepatitis B virus replication [52]. It remains to be seen if bifunctional siRNA in the skin, lung or vaginal setting is useful.
RNAi-mediated Adaptive Immune Responses
Much less research has been undertaken in the use of RNAi to modulate adaptive immune responses. Most efforts have concentrated on the ability to directly down-modulate proteins that suppress normal T cell activation such as SOCS1 or FAS ligand [53]. We have shown that RNAi can be used to elicit antigen-specific responses such that both RNAi-treated and untreated tumors are eliminated in vivo [54]. This stems from findings that tumors treated with shRNAs targeted to HPV E6/E7 downstream of the single CTL epitope were eliminated while shRNAs targeted before the epitope were not, despite each shRNA being equally as effective at gene silencing. This response was due to RNAi-mediated cleavage such that the 5’ end of the mRNA was translated and produced a truncated protein. This was rapidly degraded via the proteosome and despite overall target protein levels dropping by 60%, resulted in increased CTL presentation of the cell surface. Even non-related tumors engineered to express the target antigen were eliminated. Our work suggests that the fate of RNAi-cleaved mRNA is neither rapid nor undergo complete degradation. Moreover it suggests the possibility that appropriate targeting of genes towards the 3’end will allow for antigen specific recognition in the tumor setting.
Future Prospects
While bifunctional siRNAs hold promise as a new generation of RNA-based medicines, much development is required before this can be realized clinically. The biggest challenge with siRNA-based therapy remains delivery. Lipid-based delivery vectors are currently the preferred method for their ability to deliver siRNAs to endosomes, activating TLRs and subsequently immune responses. In the case of bifunctional siRNAs, sufficient amount of siRNAs must be delivered to target cells to cause activation of immune response as well as efficient gene silencing without causing unwanted side-effects such as toxicity. It is not clear if local or systemic delivery would be best for such effects as suppressive immune effects occur within the tumor itself. Studies by Poeck et al and Kortylewski et al utilizing lipid-based vectors to successfully deliver bifunctional siRNAs either systemically or locally have shown both approaches induce dual action resulting in superior anti-tumor effects [16, 40] but a side-by-side comparison has not been made. Clearly, adding targeting ligands in order to deliver siRNAs to specific cells will improve systemic delivery approaches [55-57]. While such studies showed significant anti-tumor effects, it would be beneficial to promote tumor-specific recognition via RNAi. This could be achieved as outlined above or perhaps via triggering some tumor cell apoptosis to recruit NK cells and regulatory T cells to the tumor site. Thus, one could envisage tri-functional siRNAs that are able to silence a gene of interest, activate innate immunity, and encourage specific recognition of tumor antigens. As different various components can easily be added to lipid-based vectors, addition of component such as CpG to siRNA-encapsulated liposomes, for example, could be a way forward. Such tri-therapy would pave the way for more potent RNA-based cancer and viral therapies.
References
- Fire, A., et al., Potent and specific genetic interference by double stranded RNA in Caenohabdartis elegans. Nature 1998. 391(6669): p. 806-11.
- Zamore, P.D., et al., RNAi: Double-Stranded RNA Directs the ATP-Dependent Cleavage of mRNA at 21 to 23 Nucleotide Intervals. Cell, 2000. 101(1): p. 25-33.
- Elbashir, S.M., et al., Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 2001. 411(6836): p. 494-8.
- Tuschl, T., RNA interference and small interfering RNAs. Chembiochem, 2001. 2: p. 239-245.
- Sontheimer, E.J., Assembly and function of RNA silencing complexes. Nature Reviews Molecular Cell Biology, 2005. 6(February 2005): p. 127-138.
- Tomari, Y., et al., A protein sensor for siRNA asymmetry. Science, 2004. 306(5700): p. 1377-80.
- Filipowicz, W., RNAi: The Nuts and Bolts of the RISC Machine. Cell, 2005. 122(1): p. 17-20.
- Dykxhoorn, D.M. and J. Lieberman, Knocking down Disease with siRNAs. Cell, 2006. 126(2): p. 231-235.
- Chalk, A., C. Wahlestedt, and E. Sonnhammer, Improved and automated prediction of effective siRNA. Biochemical and Biophysical Research Communications, 2004. 319(1): p. 264-74.
- Miyagishi, M. and K. Taira, siRNA becomes smart and intelligent. Nature Biotechnology, 2005. 23(8): p. 946-7.
- Dykxhoorn, D. and J. Lieberman, The silent revolution: RNA interference as basic biology, research tool, and therapuetic. Annual Review of Medicine, 2005. 56: p. 401-23.
- Munger, K., et al., The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol., 1989. 63(10): p. 4417-4421.
- Alexopoulou, L., et al., Recognition of double-stranded RNA and activation of NF-[kappa]B by Toll-like receptor 3. Nature, 2001. 413(6857): p. 732-738.
- Sledz, C.A., et al., Activation of the interferon system by short-interfering RNAs. Nature Cell Biology, 2003. 5(9): p. 834-39.
- Kariko, K., et al., Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J Immunol, 2004. 172(11): p. 6545-9.
- Poeck, H., et al., 5[prime]-triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med, 2008. 14(11): p. 1256-1263
- Clemens, M.J., PKR--a protein kinase regulated by double-stranded RNA. Int J Biochem Cell Biol, 1997. 29(7): p. 945-9
- Kawai, T. and S. Akira, The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol, 2010. 11(5): p. 373-384.
- Ablasser, A., et al., Selection of Molecular Structure and Delivery of RNA Oligonucleotides to Activate TLR7 versus TLR8 and to Induce High Amounts of IL-12p70 in Primary Human Monocytes. J Immunol, 2009. 182(11): p. 6824-6833.
- Bell, J.K., et al., The dsRNA binding site of human Toll-like receptor 3. Proceedings of the National Academy of Sciences, 2006. 103(23): p. 8792-8797.
- Gantier, M.P., et al., Rational Design of Immunostimulatory siRNAs. Mol Ther, 2010.
- 22. Khairuddin, N., et al., siRNA-induced immunostimulation through TLR7 promotes antitumoral activity against HPV-driven tumors in vivo. Immunol Cell Biol, 2011.
- Honda, K., et al., IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature, 2005. 434(7034): p. 772-777.
- Janeway, C.A., et al., Immunobiology (the immune system in health and disease). Vol. 6. 2005, New York: Garland Science Publishing.
- Elbashir, S.M., et al., Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J, 2001. 20(23): p. 6877-88.
- Siolas, D., et al., Synthetic shRNAs as potent RNAi triggers. Nature Biotechnology, 2005. 23(2): p. 227-231
- Rose, S.D., et al., Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucl. Acids Res., 2005. 33(13): p. 4140-4156.
- Kortylewski, M., et al., Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res, 2009. 69(6): p. 2497-505.
- Heil, F., et al., Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science, 2004. 303(5663): p. 1526-1529.
- Judge, A.D., et al., Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nature Biotechnology, 2005. 23: p. 457-462
- Vajdic, C.M. and M.T. van Leeuwen, What types of cancers are associated with immune suppression in HIV? Lessons from solid organ transplant recipients. Curr Opin HIV AIDS, 2009. 4(1): p. 35-41.
- Shankaran, V., et al., IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature, 2001. 410(6832): p. 1107-11.
- Zitvogel, L., O. Kepp, and G. Kroemer, Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol, 2011. 8(3): p. 151-60.
- Anz, D., et al., Immunostimulatory RNA blocks suppression by regulatory T cells. J Immunol, 2010. 184(2): p. 939-46.
- Akira, S., K. Takeda, and T. Kaisho, Toll-like receptors: critical proteins linking innate and acquired immunity. Nature Immunology, 2001. 2: p. 675-680.
- Beutner, K.R., et al., Imiquimod, a Patient-Applied Immune-Response Modifier for Treatment of External Genital Warts. Antimicrob. Agents Chemother., 1998. 42(4): p. 789-794.
- Krieg, A.M., Development of TLR9 agonists for cancer therapy. J Clin Invest, 2007. 117(5): p. 1184-94.
- Cheever, M.A., Twelve immunotherapy drugs that could cure cancers. Immunol Rev, 2008. 222: p. 357-68.
- Adams, S., Toll-like receptor agonists in cancer therapy. Immunotherapy, 2009. 1(6): p. 949-64.
- Kortylewski, M., et al., In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol, 2009. 27(10): p. 925-32.
- Pan, X., et al., Antitumor activity of G3139 lipid nanoparticles (LNPs). Mol Pharm, 2009. 6(1): p. 211-20.
- Aricò, E., et al., Humoral Immune Response and Protection from Viral Infection in Mice Vaccinated with Inactivated MHV-68: Effects of Type I Interferon. Journal of Interferon & Cytokine Research, 2002. 22(11): p. 1081-1088.
- Baccala, R., et al., TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med, 2007. 13(5): p. 543-551.
- Dubrot, J., et al., Intratumoral injection of interferon-alpha and systemic delivery of agonist anti-CD137 monoclonal antibodies synergize for immunotherapy. International Journal of Cancer, 2010. 9999(9999): p. NA.
- Theofilopoulos, A.N., et al., TYPE I INTERFERONS IN IMMUNITY AND AUTOIMMUNITY. Annual Review of Immunology, 2005. 23(1): p. 307-335.
- Wong, T., et al., Therapeutic implications for interferon-alpha in arthritis: a pilot study. J Rheumatol, 2003. 30(5): p. 934-40.
- Hui, C.K., et al., Interferon and ribavirin therapy for chronic hepatitis C virus genotype 6: a comparison with genotype 1. J Infect Dis, 2003. 187(7): p. 1071-4.
- Berkhout, B. and O. ter Brake, Towards a durable RNAi gene therapy for HIV-AIDS. Expert Opinion on Biological Therapy, 2009. 9(2): p. 161-170.
- Arbuthnot, P., et al., Opportunities for treating chronic hepatitis B and C virus infection using RNA interference. Journal of Viral Hepatitis, 2007. 14(7): p. 447-459
- DeVincenzo, J., et al., Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV). Antiviral Res, 2008. 77(3): p. 225-31.
- Nguyen, D.N., et al., Drug Delivery-mediated Control of RNA Immunostimulation. Mol Ther, 2009.
- Hornung, V., et al., Sequence-specific potent induction of IFN-[alpha] by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med, 2005. 11(3): p. 263-270.
- Dotti, G., et al., Human cytotoxic T lymphocytes with reduced sensitivity to Fas-induced apoptosis. Blood, 2005. 105(12): p. 4677-84.
- Gu, W., et al., Both treated and untreated tumors are eliminated by short hairpin RNA-based induction of target-specific immune responses. Proceedings of the National Academy of Sciences, 2009: p. -.
- Podesta, J.E. and K. Kostarelos, Chapter 17 Engineering Cationic Liposome: siRNA Complexes for In Vitro and In Vivo Delivery, in Methods in Enzymology, D. Nejat, Editor. 2009, Academic Press. p. 343-354.
- Sasaki, A. and M. Kinjo, Monitoring intracellular degradation of exogenous DNA using diffusion properties. Journal of Controlled Release, 2010. 143(1): p. 104-111.
- Kim, S.-S., et al., Targeted Delivery of siRNA to Macrophages for Anti-inflammatory Treatment. Mol Ther, 2010. 18(5): p. 993-1001.
Associate Professor Nigel McMillan is a Principal Research Fellow at the University of Queensland Diamantina Institute. His research investigates RNA interference for the treatment of viral diseases and cancers and has published in the area of RNAi delivery, RNAi and the immune system.
Dr. Norliana Khairuddin is a graduate from the McMillan Laboratory and pioneered the studies into immunostimulatory siRNAs for the treatment of cervical cancer.
This article was printed in the 12/20/2011 issue of International Drug Discovery,6,, 6,. Copyright rests with the publisher.